About this course:
The purpose of this module is to examine the pathophysiology of normal blood clotting as well as the most common disorders of excessive blood clotting and bleeding, exploring the clinical manifestations and nursing implications of each disease process to enhance nursing practice and safeguard patient care.
Course preview
The purpose of this module is to examine the pathophysiological basis of normal blood clotting as well as the most common disorders of excessive blood clotting and bleeding, exploring the clinical manifestations and nursing implications of each disease process to enhance nursing practice and safeguard patient care.
By the completion of this module, the nurse should be able to:
- Describe the pathophysiology of normal bleeding and blood clotting, discuss the distinction between primary and secondary hemostasis, as well as provide an overview of the clotting cascade and coagulation factors.
- Describe the pathologic mechanisms of blood clotting failure or impairment, delineating the most common domains of dysfunction.
- Review the most common disorders of excessive blood clotting, inheritance patterns, and clinical manifestations, and briefly discuss anticoagulation therapy with regards to its role in managing these disorders, highlighting the pertinent aspects of nursing care and key patient teaching points.
- Review the most common bleeding disorders, inheritance patterns, and their clinical manifestations, highlighting the pertinent aspects of nursing care, monitoring, and key patient teaching points.
When the body's physiologic mechanisms of blood clotting fail, excessive blood clotting or excessive bleeding can ensue. Disorders of blood clotting occur when there is an imbalance between factors that promote blood clotting (procoagulants) and the factors that inhibit blood clotting (anticoagulants). Blood clotting disorders are characterized by an impairment in the ability to effectively form a blood clot (or thrombus) in response to an injury. They may also be due to a hypercoagulable state in which there is an increased tendency to develop thrombus' in the arteries and veins. These conditions can be inherited (congenital) due to specific genetic defects present at birth, or acquired (developed), resulting from an underlying medical condition, trauma, surgery, or medication. If not promptly identified and effectively treated, blood clotting disorders can lead to serious health consequences, including hemorrhage, heart attack, stroke, organ failure, and death. To understand blood clotting disorders, it is first essential to understand the pathophysiologic basis of blood cell production, and the mechanisms of bleeding and blood clotting (Longo, 2019).
Hematopoiesis
Hematopoiesis is the ongoing process of blood cell production in the human body that primarily occurs in the bone marrow. The process is regulated through a series of steps in which undifferentiated cells are biochemically stimulated to undergo mitotic cell division (i.e., proliferation) and cell maturation (i.e., differentiation). Hematopoiesis continues throughout the lifespan, functioning to maintain homeostasis within the body in response to infection or injury (Longo, 2019). When there is an increase in the destruction of circulating cells, such as during an acute bleeding event, hematopoiesis accelerates to generate more cells and compensate for the loss. In long-term dysfunction, as in chronic illness, there is a more significant increase in hematopoiesis than in acute conditions such as hemorrhage. As demonstrated in Figure 1, every cell type in the body originates from hematopoietic stem cells. The stem cells grow, multiply, and differentiate under the control of cytokines and growth factors. During the differentiation process, the stem cells follow distinctive paths to maturity, and travel down committed lines of blood cells, primed to perform a specific function. The average human body requires nearly 100 billion new blood cells per day, which is why hematopoietic stem cells are self-renewing or can proliferate by themselves so that a relatively constant population of stem cells is always readily available (McCance & Heuther, 2019).

Components of Blood
Blood is a vital component of the body. It serves several important functions, including transporting oxygen and nutrients to tissues within the body, regulating temperature, and responding to blood vessel injuries to maintain homeostasis. The amount of blood within each individual depends on their size, but the average adult has approximately 5 liters (1.3 gallons) of blood circulating within their body. Blood consists of both solid (formed) elements (white blood cells [WBCs], red blood cells [RBCs], and platelets), and liquid components (plasma) (Longo, 2019).
WBCs
WBCs are the components of the immune system which work to fight infection and other illnesses. WBCs are comprised of five specific subtypes (neutrophils, monocytes, macrophages, eosinophils, and basophils). Each WBC serves a specific function in mediating the inflammatory and immune response to infection. WBCs have variable lifespans, as some may live for only 24 hours, but the average WBC lifespan is 13 to 20 days (Longo, 2019).
RBCs
RBCs are mature red blood cells (erythrocytes) and carry hemoglobin, a protein that transports oxygen from the lungs to all the tissues within the body. The body relies on oxygen as a critical component for all cellular functioning and processes. Hemoglobin also carries waste products (mainly carbon dioxide) from the tissues back to the lungs, where waste is expelled through breathing. Erythrocytes comprise about 45% of total blood, have an average lifespan of 120 days, and are pink in appearance due to their high hemoglobin content. Hematocrit reflects the percentage of RBCs in a given volume of blood (Longo, 2019).
Platelets
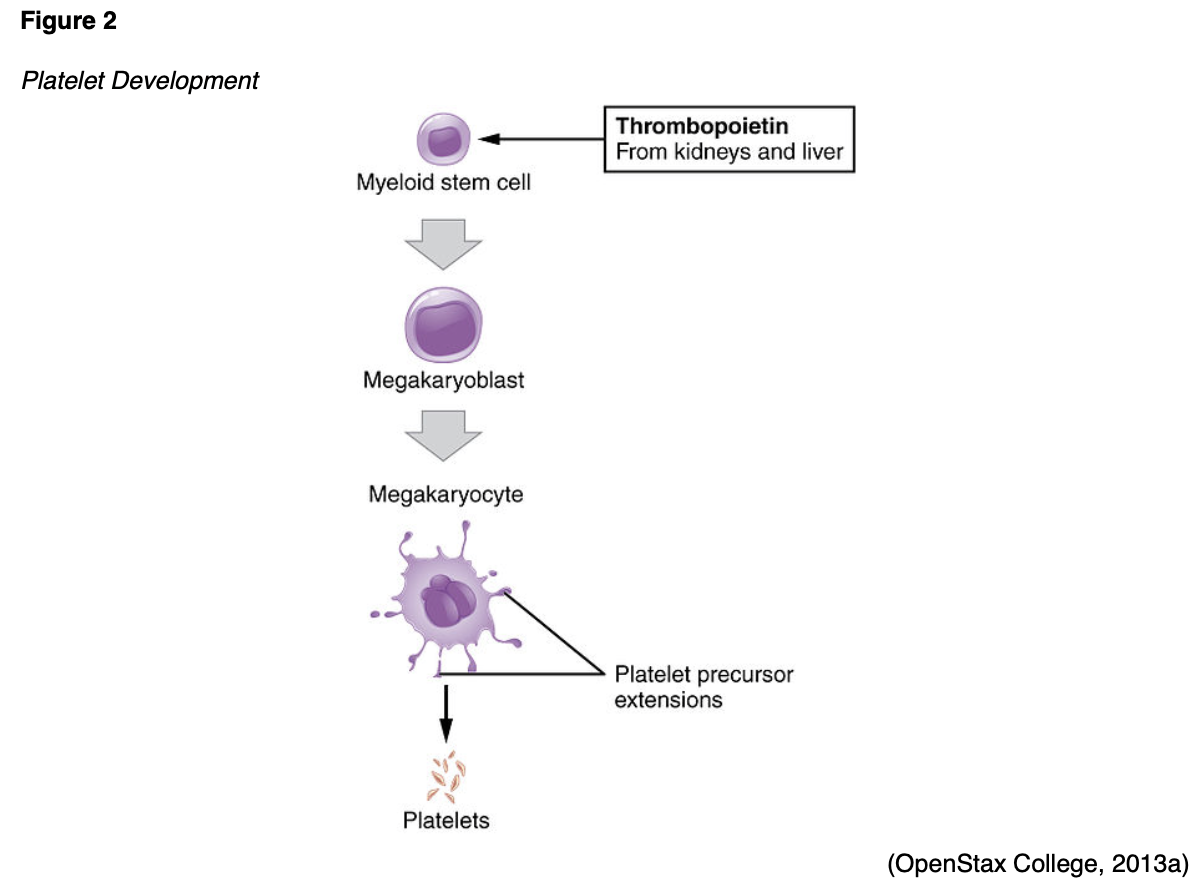
Platelets, also called thrombocytes, have a critical role with regards to all aspects of bleeding and clotting. They are produced in the bone marrow from cells called megakaryocytes. The process of platelet differentiation, as outlined in Figure 2, is regulated under the control of thrombopoietin (TPO), a hormone produced by the liver and kidneys. TPO influences the growth and differentiation of myeloid stem cells into megakaryoblasts, the precursor cell to the promegakaryocyte, which, in turn, develops into a giant megakaryocyte. As demonstrated earlier in Figure 1, megakaryocytes are depicted as notably larger than all the other cells because each megakaryocyte is prepared to produce and release more than 1,000 platelets, accounting for their large size. Platelets are colorless fragments of blood cells that are essential for blood clotting in response to an injury. They contain proteins on their surfaces that allow them to stick to each other and blood vessel walls. Their primary function is to gather at the site of the blood vessel injury to seal small cuts or breaks in blood vessels to control bleeding. They function in collaboration with proteins called clotting factors to stop bleeding and change shape in response to injury to ensure all injured parts of the vessel are sealed. Platelets have an average lifespan of 7 to 10 days, and a healthy adult platelet count is 150,000–450,000/microliter (μL). Thrombocytopenia is a condition where the platelet count is lower than normal, thereby heightening the risk for bleeding events and hemorrhage. Thrombocytosis occurs when there is an excessive number of platelets in the blood, increasing the risk of forming blood clots (Longo, 2019).
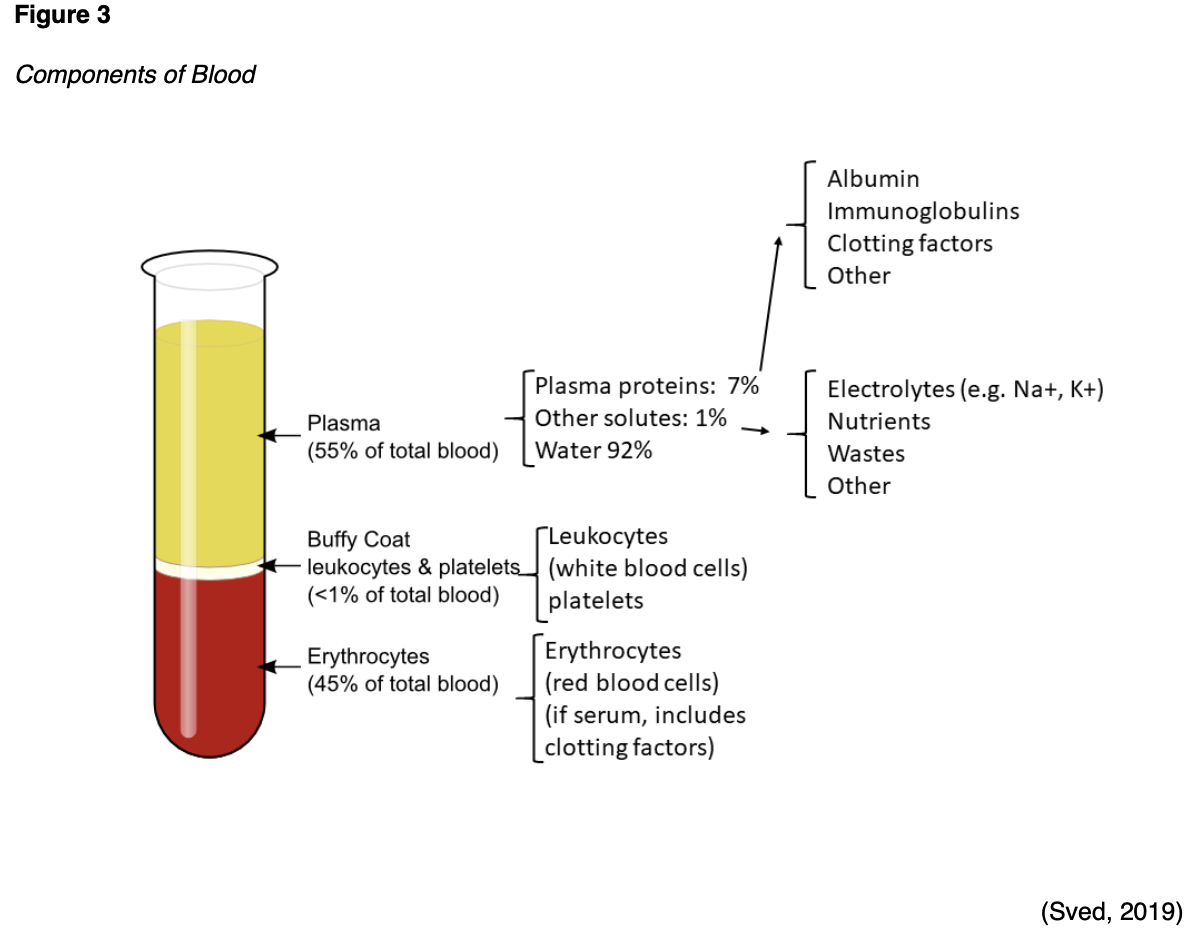
Plasma
Plasma is an aqueous component of blood that functions to carry nutrients, proteins, and hormones throughout the body and transport waste products to the kidneys and digestive tract for removal. As demonstrated in Figure 3, plasma accounts for about 55%
...purchase below to continue the course
Physiologic Mechanisms of Blood Clotting (Coagulation)
Blood clotting, commonly referred to as coagulation, is a vital mechanism that protects against blood loss due to bleeding. A complex interplay mediates the process between vascular mechanisms, enzymes, hormones, proteins, and cells. Hemostasis, or the cessation of bleeding, refers to the body's physiological response to stop bleeding from an injured blood vessel to prevent blood loss. The coagulation process is divided into two stages: primary hemostasis (which results in the formation of a weak platelet plug) and secondary hemostasis (which stabilizes the weak platelet plug, evolving into a secure and insoluble fibrin clot). The defining components of each process are as follows:
- Primary hemostasis:
Vascular spasm or vasoconstriction of the blood vessel,
Platelet adhesion,
Platelets activation (or secretion),
Platelet aggregation.
- Secondary hemostasis:
Activation of clotting factors,
Conversion of prothrombin to thrombin,
Conversion of fibrinogen to fibrin (Garmo et al., 2020).
As outlined on the left side of Figure 4 (part a), upon recognition of an injury, the vessel constricts (tightens) to reduce blood flow to the area to prevent further blood loss. Vasoconstriction is mediated by endothelin-1, a potent vasoconstrictor, which is synthesized by the damaged endothelium. The damaged endothelium leaks collagen and proteins, including von Willebrand factor (vWF), into the bloodstream. vWF, a protein, is the primary mediating factor for platelet adhesion, acting like a glue and binding the platelets with enough strength to withstand stress and prevent detachment. Platelets circulating in the blood travel to the site of injury and adhere to the vessel walls, sticking together to form a temporary platelet plug. Platelets mainly bind to collagen in tissues at the site of vessel wall injury and exposed vWF at the damaged vessel site. When this happens, signals are sent into the bloodstream, calling more platelets to the site of injury; inducing platelet activation. Platelet aggregation begins once platelets have been activated. Calcium is released and attaches to the phospholipids (a main component of cellular membranes), which appear secondary to the platelet activation, offering a surface for various coagulation factors. Thromboxane A2, a potent vasoconstrictor, is formed during platelet activation; it functions as a stimulus for platelet aggregation and mediates the release of adenosine phosphate (ADP). ADP is a platelet agonist that activates other platelets, directing more platelets to the injury area. ADP enables platelets to clump together and change shape as a means to increase their surface area. Thrombin, formed during the coagulation cascade (which will be reviewed in the next section), is the most potent activator of platelets and is considered the key enzyme of coagulation. Fibrinogen, along with the platelets, creates a weak platelet plug that temporarily protects against further bleeding (Gross et al., 2018). Secondary hemostasis subsequently kicks in, and the clotting factors act to stabilize the platelet plug and convert it into a hard, insoluble fibrin clot (Garmo et al., 2020).

Clotting Factors
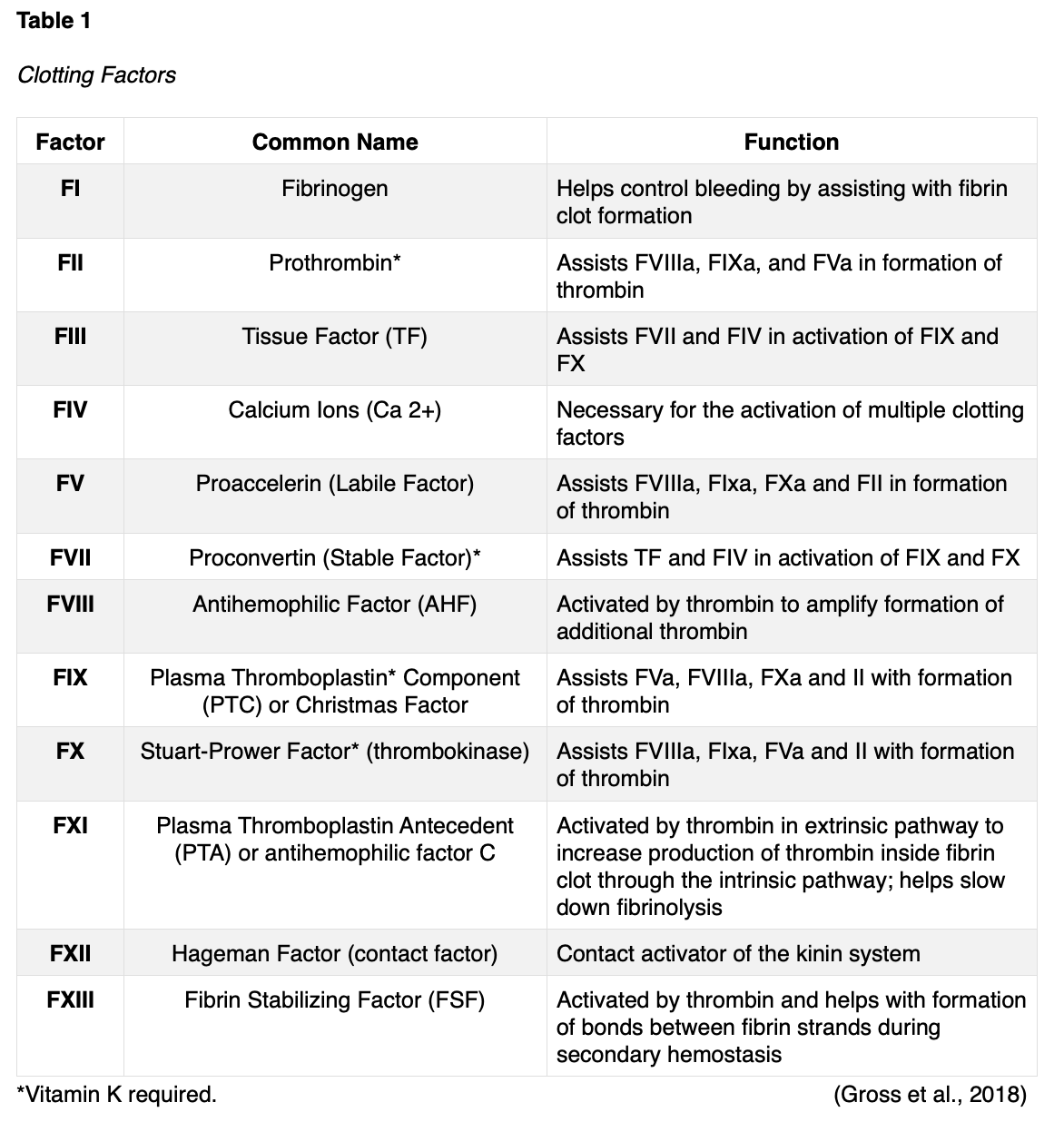
There are 12 clotting factors (F) labeled in roman numerals (FI through FXIII) as outlined in Table 1. Of note, there is no clotting FVI. Clotting factors are proteins that are primarily secreted by the platelets and liver and are dependent upon calcium ions (Ca2+), and vitamin K. Ca2+ is vital for the acceleration of blood clotting. Vitamin K is a naturally occurring fat-soluble vitamin synthesized by bacteria residing in the large intestine and is also consumed in the diet. Vitamin K assists with blood clotting, bone metabolism, regulation of blood calcium levels, and is required to produce prothrombin (FII). Therefore, vitamin K deficiencies can lead to bleeding and hemorrhage, bone mineralization, and osteoporosis. Ca2+, otherwise referred to as FIV, is derived from the breakdown of bone and is also acquired via the diet (McCance & Heuther, 2019).
Coagulation Cascade
The coagulation cascade, as demonstrated in Figure 4 (part b), is activated through two distinct yet interconnected pathways (intrinsic and extrinsic) that converge into the common pathway, which lead to the formation of a fibrin clot. All three pathways are mediated by hormones, proteins, and clotting factors through a series of complex events. The ultimate goal of the coagulation cascade is to convert fibrinogen (F1), a soluble plasma protein into fibrin, a non-soluble plasma protein. The coagulation cascade can be summarized by the following three essential steps:
- Extrinsic and intrinsic pathways form the enzyme prothrombinase;
- Prothrombinase converts prothrombin to thrombin;
- Thrombin converts fibrinogen into fibrin (Gross et al., 2018).
Extrinsic pathway. Clotting factors involved in the extrinsic pathway include FVII and FIII. The beginning of the process towards fibrin clot formation is carried out by the extrinsic pathway, activated in response to external tissue trauma, in which blood escapes from the vascular system. Once the damage is identified, tissue factor (TF, or FIII), a cell membrane protein, is released from the damaged cells. TF binds with prothrombin (FVII), activating it into FVIIa, thereby initiating the clotting cascade. The extrinsic pathway is much quicker and less complex than the intrinsic pathway.
Intrinsic pathway. Clotting factors involved in this pathway include FXII, FXI, FIX, and FVIII. The intrinsic pathway is activated by trauma that begins inside the vascular system, prompted by internal damage to the vessel wall. FXII is activated into FXIIa when it comes in contact with the damaged endothelial collagen, thereby igniting the intrinsic pathway. This pathway is slower than the extrinsic pathway but is considered more important.
Common Pathway. Clotting factors involved in the common pathway include FX, FV, FII, FI, and FXIII. The extrinsic and intrinsic pathways converge into the common pathway with the activation of FX into FXa, which subsequently combines with FVa and Ca2+ on phospholipid surfaces. This process activates FII into thrombin, which then activates FXIIIa. FXIIIa interacts with fibrin to form the stabilized clot (Garmo et al., 2020; Gross et al., 2018).
Antithrombotic Mechanisms
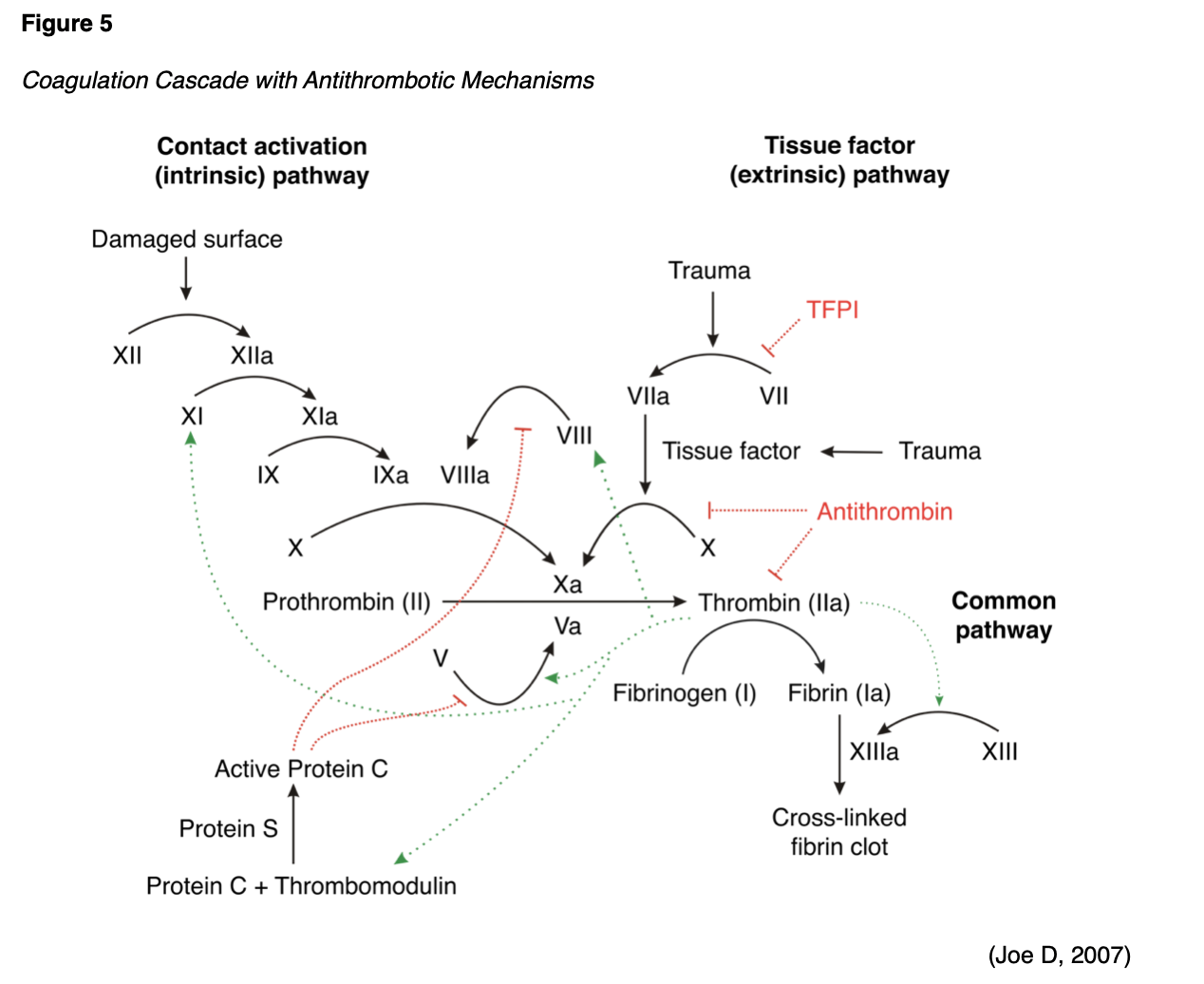
Parallel to the coagulation cascade, the body also has an innate, highly regulated process for controlling and preventing abnormal blood clot development. Antithrombotic mechanisms are naturally occurring anticoagulants in the body. An anticoagulant refers to any substance that opposes coagulation. Coagulation is regulated by several antithrombotic mechanisms to preserve the fluidity of the blood and limit blood clotting to vascular injury sites, so blood clots do not form under normal conditions. The endothelial cells provide several antithrombotic effects. They produce substances that act to inhibit platelet binding, secretion, and aggregation, and generate anticoagulant factors such as tissue factor pathway inhibitor (TFPI) and antithrombin. Various circulating plasma anticoagulants instinctively function to limit the coagulation process to the area of injury and to reestablish and maintain patency of blood vessels. Endothelial cells also activate the fibrinolytic mechanisms through the production of tissue plasminogen activator 1 (tPA) and plasminogen activator inhibitor. Further, basophils of the immune system also play a fundamental role in preventing blood clotting through their release of heparin, a short-acting anticoagulant that opposes prothrombin. Heparin is additionally found on the surfaces of cells lining the blood vessels and promotes blood fluidity. The major physiologic antithrombotic pathways and their impact on the coagulation cascade are illustrated in Figure 5 (highlighted in red) and described below (Garmo et al., 2020; Gross et al., 2018).
Antithrombin (AT)
Previously referred to as antithrombin III, AT is the major inhibitor of thrombin and other clotting factors in the coagulation cascade. AT is activated by binding to heparin on endothelial cell surfaces. It binds to and inactivates FIXa, FXa, FXIa, and FXIIa, and opposes the conversion of prothrombin (FII) to thrombin in the common pathway. AT neutralizes thrombin and other activated clotting factors on the vascular surface (Longo, 2019).
Protein C and Protein S (Protein C/S)
Protein C/S refers to a cluster of plasma proteins that work together to regulate blood clot formation. Protein C inactivates clotting factors involved in the intrinsic pathway, serving as a natural anticoagulant when activated by thrombin. Thrombin-induced activated protein C (APC) occurs on thrombomodulin, a transmembrane binding site for thrombin on the endothelial cell surface. APC subsequently inactivates FVa and FVIIIa, slowing down blood clot formation. Protein S exists in two forms; free and bound to another protein. As demonstrated in Figure 5, free protein S is a cofactor to protein C, functioning to accelerate the inactivation of FVa and FVIIIa (Longo, 2019).
Tissue Factor Pathway Inhibitor (TFPI)
TFPI is generated primarily by endothelial cells and inhibits the TF/FVIIa/FXa complex, turning off the initiation of the coagulation cascade's extrinsic pathway. TFPI is bound to lipoprotein and can be released by heparin. It also circulates in plasma in a free form in small quantities, acting as a physiologically active anticoagulant (Longo, 2019).
Fibrinolytic System
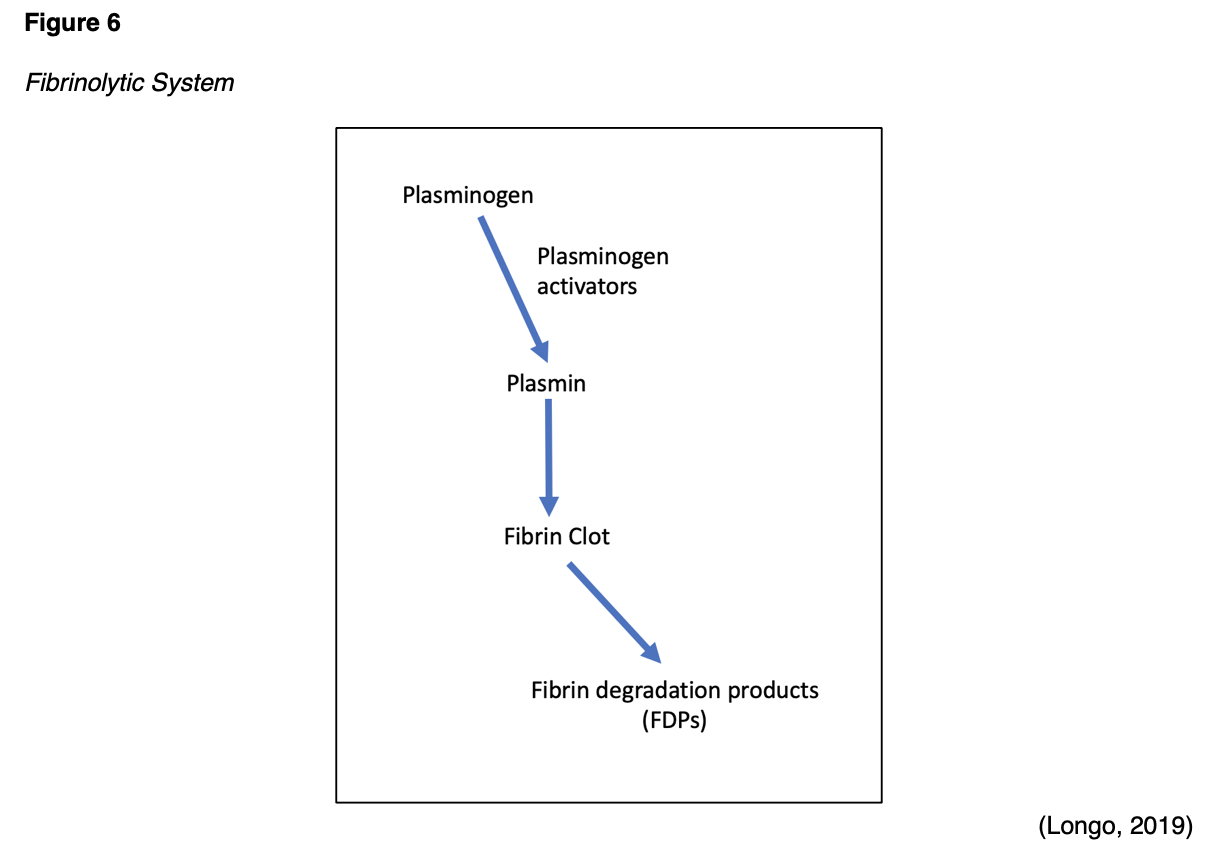
Any thrombin that evades the inhibitory effects of the body's natural anticoagulant system is used to convert fibrinogen to fibrin. However, once the injured blood vessel heals, the clot must eventually be removed to reestablish normal blood flow and maintain patency of the blood. Therefore, the fibrinolytic system and fibrinolysis process are activated to facilitate the breakdown and removal of these unwanted fibrin deposits. In addition to the degradation of fibrin, fibrinolysis also regulates the abnormal development of blood clots and manipulates the extent of blood clot formation. Plasmin is the primary enzyme of the fibrinolytic system, functioning to digest fibrin into fibrin degradation products (FDPs). Plasmin is formed through a series of reactions, as demonstrated in Figure 6. Tissue plasminogen activator (tPA) is released from endothelial cells and binds to the fibrin clot. Subsequently, inactivated plasminogen (an enzyme that degrades fibrin clots) is converted into active plasmin. The process of fibrinolysis can be primary or secondary. Primary fibrinolysis is the body's natural process to maintain homeostasis, as described above. In contrast, secondary fibrinolysis refers to the dysfunctional breakdown of blood clots secondary to medical disorders, infections, or medications, and can induce severe bleeding (Garmo et al., 2020; Leung, 2020; Longo, 2019).
Thrombosis
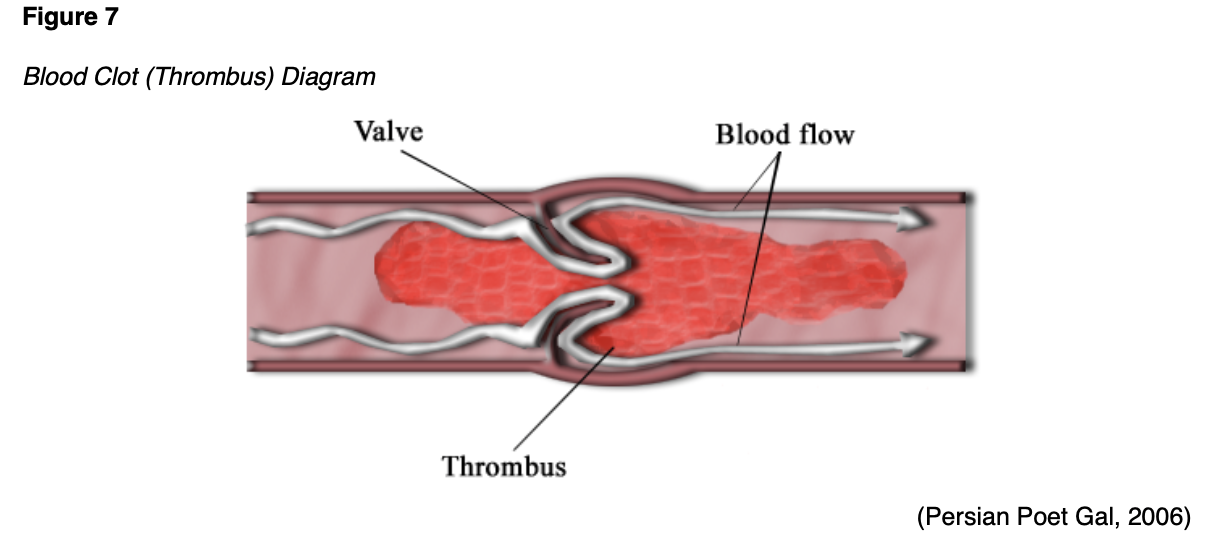
Thrombosis is a pathologic process in which there is an increased tendency to develop abnormal blood clots (thrombi) within uninjured blood vessels. A thrombus is a stationary clot attached to the vessel wall primarily composed of fibrin and other blood cells. As demonstrated in Figure 7, the thrombus can grow large enough to reduce or obstruct blood flow within the vessel, thereby depriving tissues and organs of vital nutrients, inducing tissue ischemia and hypoxia (Longo, 2019).
Virchow’s Triad
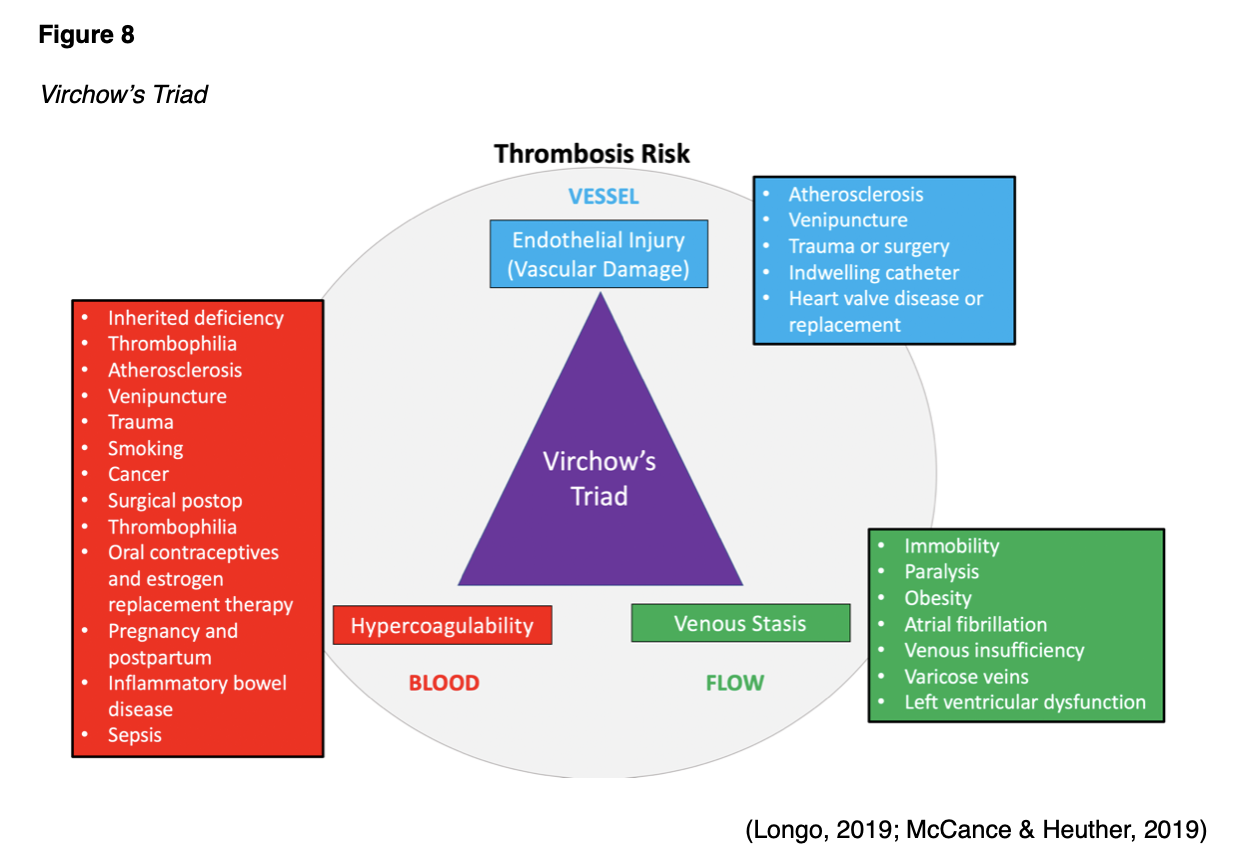
Rudolf Virchow, often referred to as the father of modern pathology, is a 19th Century physician credited with identifying three significant predisposing abnormalities that can lead to the development of thrombosis. Branded "Virchow's triad," the three pathologic mechanisms influencing blood clot formation are listed below and explained within Figure 8:
- Damage to the blood vessel wall;
- Blood flow abnormalities;
- Hypercoagulability of the blood (McCance & Heuther, 2019)
Endothelial Injury
As described earlier, vascular endothelial injury under physiologic conditions leads to platelet activation and blood clot formation to prevent blood loss and restore the integrity of the vascular wall. However, when these mechanisms are impaired, or there is an underlying defect, pathologic consequences can ensue. Endothelial injury can be precipitated by numerous insults that extend beyond external tissue trauma, such as smoking, surgery, atherosclerotic disease, inflammation, or chronic elevations in blood pressure. Regardless of the precipitating event, when the endothelium is injured, normal blood flow is altered, inducing "turbulence" within the vessel. Turbulence is defined as the increased rate of blood flow over the injured surface, generating disordered current and increased friction within the vessel. Atherosclerotic plaques from hyperlipidemia commonly generate turbulent environments within blood vessels (McCance & Heuther, 2019).
Venous Stasis
Venous stasis refers to the loss of blood flow within the vessel. Blood pooling is a tremendous risk factor for thrombus formation, as, during venous stasis, platelets remain in contact with the endothelium for a prolonged period. During this time, clotting factors that would usually be diluted with flowing blood become concentrated and potentially activated. As listed above in Figure 8, the most common conditions that predispose patients to venous stasis include immobility, paralysis, venous insufficiency, varicose veins, obesity, heart failure, and low cardiac output (McCance & Heuther, 2019).
Hypercoagulability
Abnormalities within the coagulation cascade, fibrinolytic pathways, and/or platelet function contribute to hypercoagulability and subsequent thrombus formation. Hypercoagulability may result from hereditary or acquired etiologies and is usually multifactorial, with lifestyle and environmental factors playing key roles. Hypercoagulability resulting from inherited defects includes Factor V Leiden (FVL), prothrombin mutation (G20210A), or deficiencies of the natural proteins that prevent clotting described within the next section of this module. Some individuals may present with increased levels of certain clotting factors without any identifiable genetic mutation. The most common causes of acquired hypercoagulability include cancer, inflammation, immobility from paralysis or prolonged bed rest, surgery (especially the immediate postoperative period), pregnancy, and the postpartum period. Certain medications are known to increase the coagulability of the blood, such as oral contraceptives and hormone replacement therapy (McCance & Heuther, 2019). Additional risk factors for thrombus formation that extend beyond those identified within Virchow’s triad include the following:
- Personal history of blood clots;
- Family history of blood clots (even without an identifiable inherited disorder);
- Hypertension (high blood pressure);
- Chronic inflammatory diseases;
- Diabetes;
- Age (increased risk in people over the age of 60);
- Presence of multiple concurrent risk factors (i.e., smoking cigarettes while taking oral contraceptive medications) (Longo, 2019).
Provoked vs. Unprovoked. Thrombi can be either provoked or unprovoked, which is essential for determining the duration of therapy. Unprovoked implies that no identifiable risk factor or underlying cause is evident or identifiable; a provoked thrombus is one that is caused by a known event, such as surgery or prolonged immobility. Unprovoked events are considered more serious and require specific diagnostic workup and treatment, usually under the care of a hematologist (Lip & Hull, 2020).
Arterial vs. Venous Thrombosis
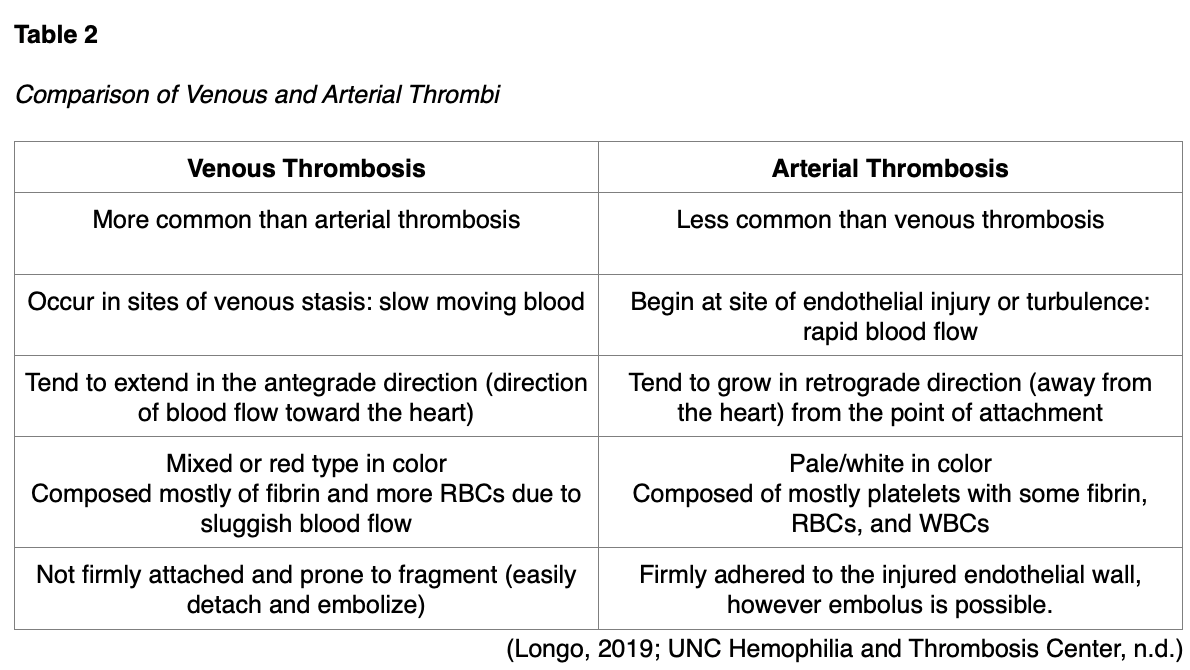
A thrombus can develop anywhere within the cardiovascular system, with the majority classified as venous (within a vein) or arterial (within an artery). Arteries are the blood vessels responsible for carrying oxygen-rich blood away from the heart to the body tissues, whereas veins are the blood vessels that carry blood back to the heart. Venous thrombosis is also called venous thromboembolism (VTE) and includes two major types; deep vein thrombosis (DVT) and superficial vein thrombosis (SVT). DVT is the most common type, arising in the large veins of a lower extremity. Arterial thrombi almost always arise from the heart (i.e., aorta, coronary arteries) but may also occur in the brain's cerebral arteries. Arterial thrombosis is commonly caused by arteriosclerosis, a condition in which a fatty substance called plaque builds up along the walls of arteries. This plaque formation hardens and narrows arteries, and the plaque can crack or rupture, attracting platelets and eventually leading to the formation of blood clots. While thrombi vary in shape and size, they all commonly maintain an area of attachment to an underlying vessel or heart wall. The distinguishing features and characteristics of venous and arterial thrombi are outlined in Table 2 (National Heart, Lung, and Blood Institute [NHLBI], n.d.b).
Complications of Venous Thrombosis
Since thrombosis can block the blood flow in both veins and arteries, complications depend on the clot's location. The most serious and dangerous complication of a VTE is a pulmonary embolism (PE), or a blood clot within the lung. A thrombus detaches from the vessel wall and circulates within the bloodstream, causing a sudden blockage of a vessel called an embolism. As demonstrated in Figure 9, a PE is most commonly caused by DVT, as a piece of the blood clot within the extremity breaks off, travels through the veins, and becomes lodged in the lung's blood vessels. A PE can completely obstruct blood flow and induce sudden death in some patients. It can also cause hypoxia or low oxygen levels in the blood, which can lead to permanent damage to the lungs or other organs in the body due to receiving insufficient oxygen (McCance & Heuther, 2019).
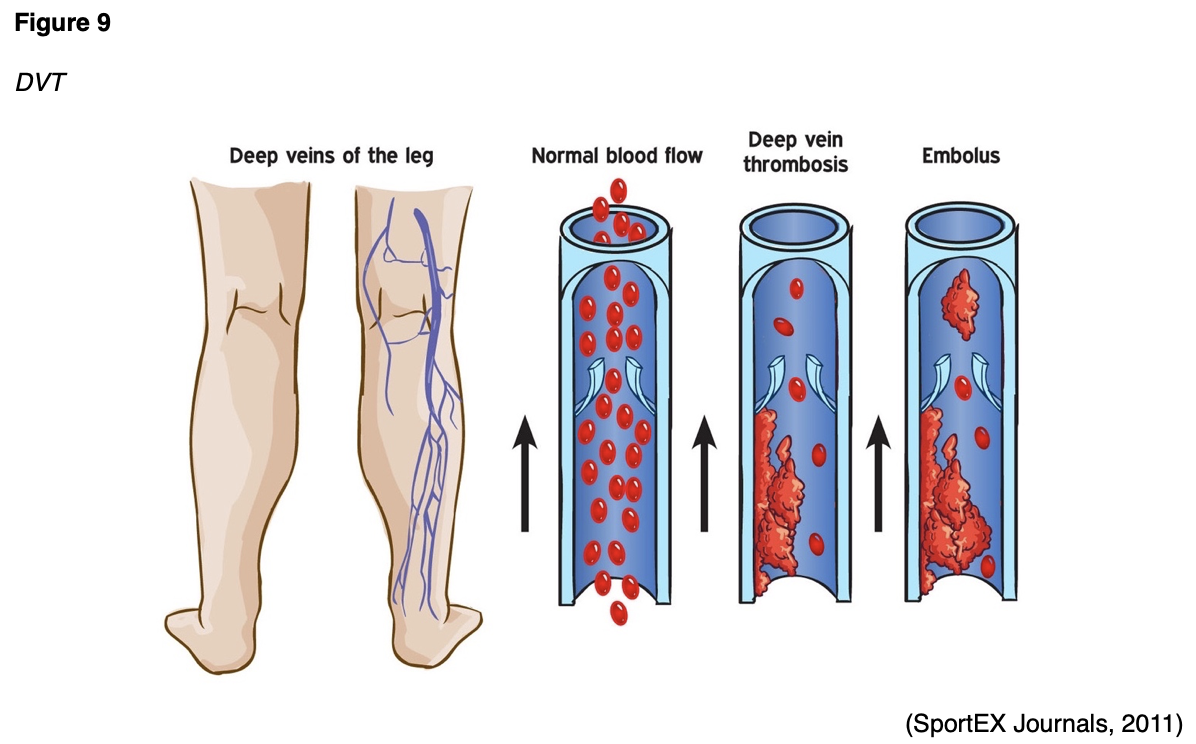
VTEs can also lead to serious long-term complications, including post-thrombotic syndrome (PTS), which is the most common complication of a DVT. PTS causes chronic pain and swelling in the affected extremity and, in severe cases, can lead to venous ulcers. Less commonly, some patients can develop chronic thromboembolic pulmonary hypertension (CTEPH) in which abnormally high blood pressure in the arteries of the lungs causes the right side of the heart to work harder than normal, leading to heart failure (McCance & Heuther, 2019).
Complications of Arterial Thrombosis
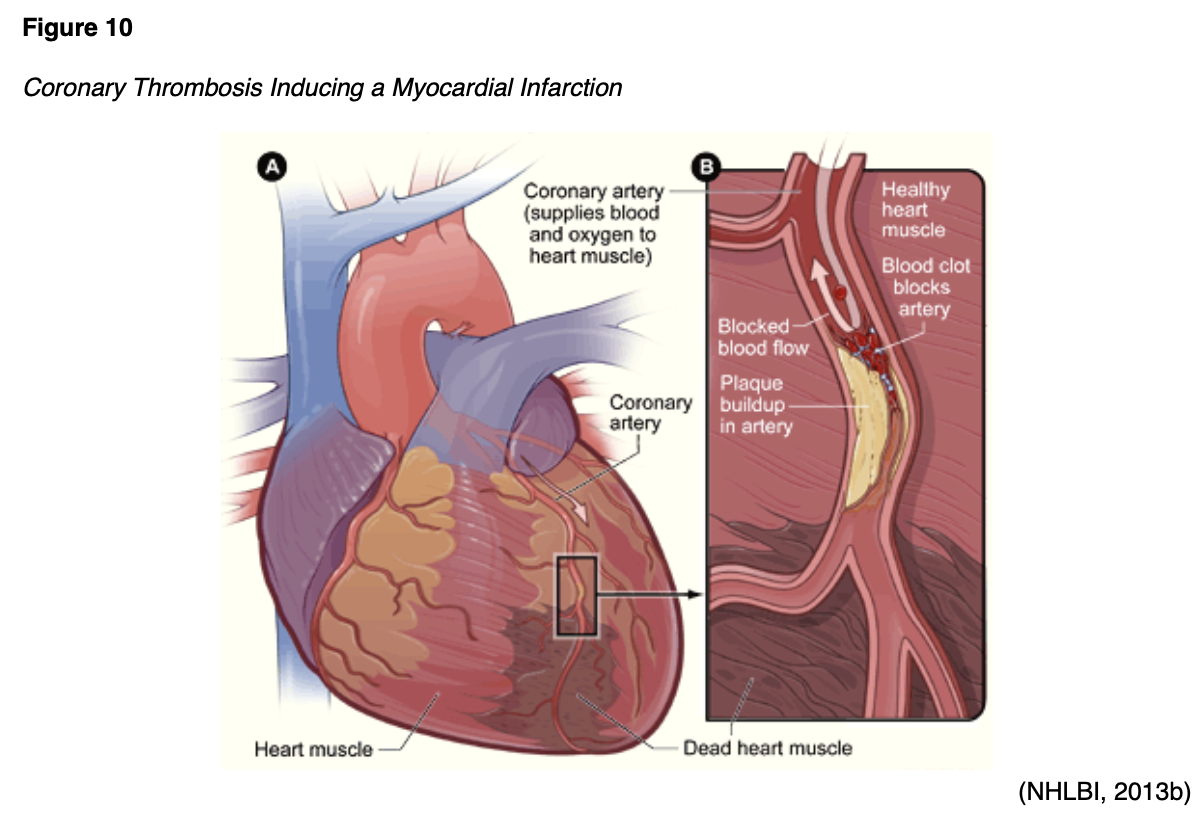
Arterial thrombosis and emboli can cause tissues to become starved of blood and oxygen, leading to necrosis and infarction of vital tissue and organs (heart, brain, kidney, spleen). As shown in Figure 10, a coronary thrombosis is a blockage that occurs within an artery of the heart and causes a myocardial infarction (MI) or heart attack (McCance & Heuther, 2019).
Arterial thrombosis can also cause an ischemic stroke, which occurs when cerebral blood flow is reduced to critical levels or stopped altogether, as demonstrated in Figure 11. An ischemic stroke can either be caused by a thrombus that originates within an artery that supplies blood to the brain, or an embolus that becomes lodged in and blocks an artery that supplies blood to the brain. In both types of ischemic stroke, the plaque or clot keeps oxygen-rich blood from getting to a portion of the brain, and neurons deprived of oxygen start to die within minutes (NHLBI, n.d.b).
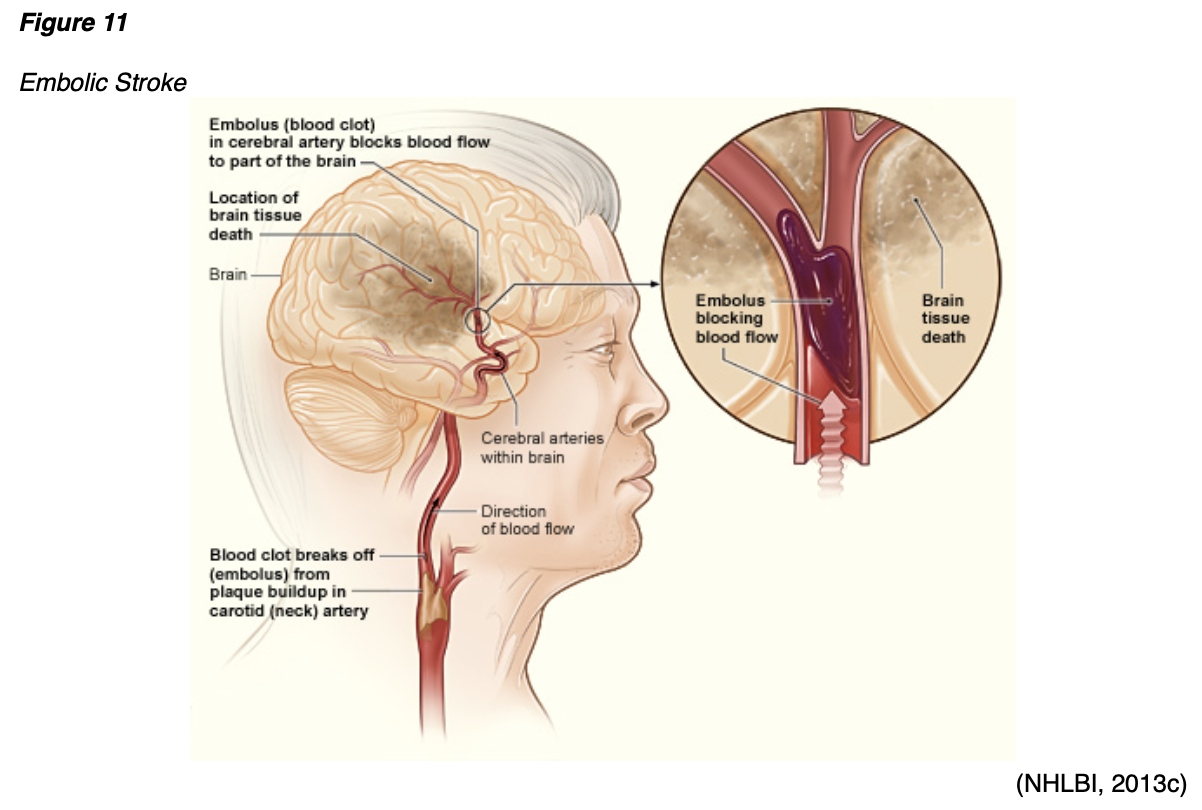
Ischemic stroke is most commonly caused by atherosclerosis but can also be due to atrial fibrillation (Afib), a type of cardiac arrhythmia. Afib is characterized by an irregular rhythm in which blood is not appropriately pumped out of the heart, leading to periods of blood pooling within the heart's atrium. This increases the risk of embolic stroke if a blood clot forms there and is then expelled and travels up to the brain, as portrayed in Figure 12 (McCance & Heuther, 2019; NHLBI, n.d.b).

Arterial emboli can also occur in the periphery, such as the legs and feet, reducing blood flow or causing acute ischemia of the lower limbs. This can occur in one of two ways; from arterial embolism or from an arterial thrombus induced by an atherosclerotic artery (atheroembolism) within the extremity, which is demonstrated in Figure 13. When emboli originate in the heart, greater than 70% will obstruct the lower limbs. The femoral artery at the femoral bifurcation is the most common site affected, accounting for 35 to 50% of all cases. Other arteries of the lower extremity commonly affected include the popliteal, iliac, and the aorta. Complete obstruction of blood flow can lead to gangrene, in which the extremity becomes cold, painful, and eventually turns black from necrosis or cell death. In these cases, patients usually require amputation (Casas, 2019).

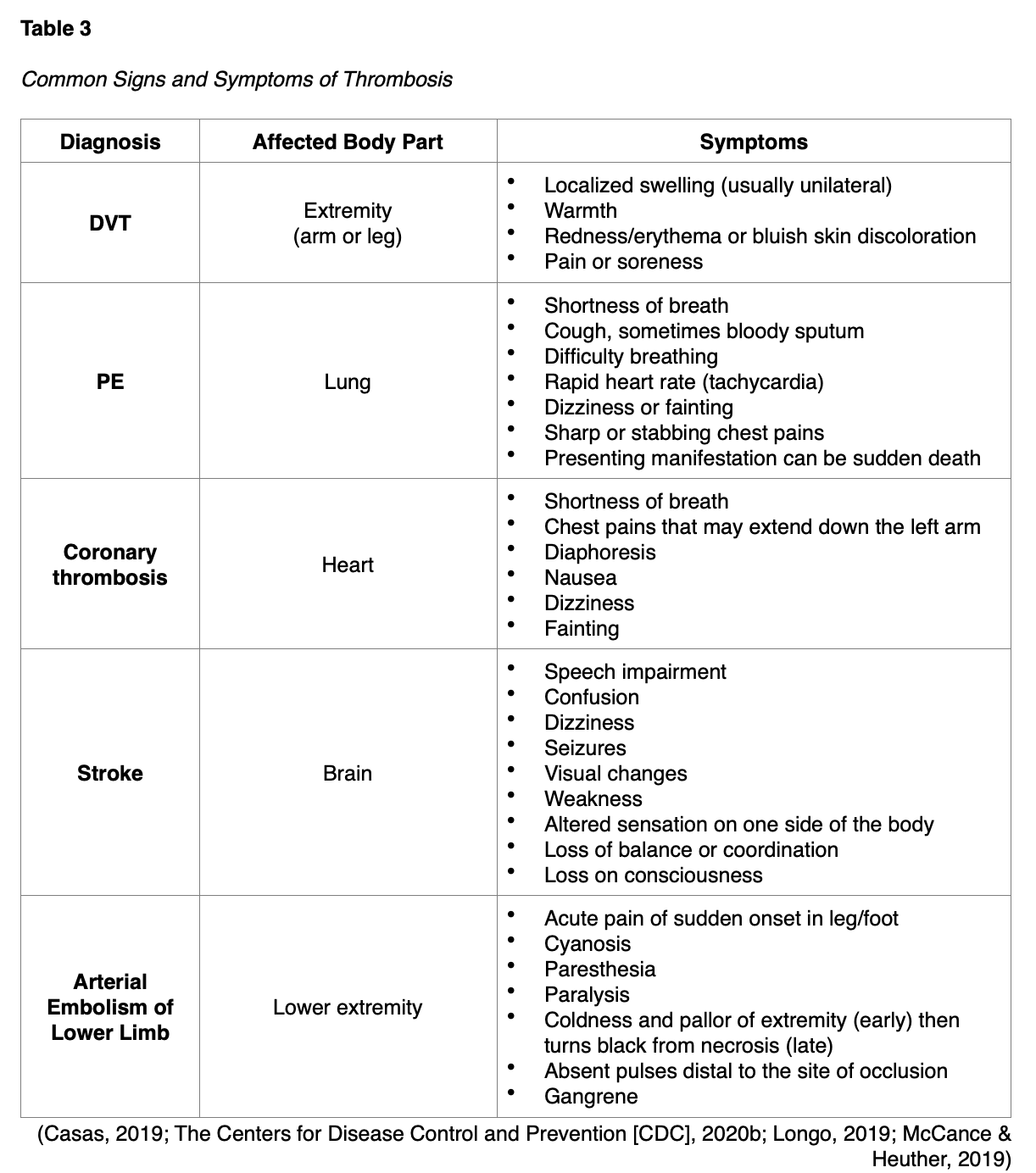
Signs and Symptoms of Thrombosis
Since thrombosis can lead to significant morbidity and mortality, resulting in serious and life-threatening consequences, early identification and intervention are essential. Signs and symptoms can vary depending upon the location of the body affected, with the most common outlined in Table 3.
Anticoagulation Therapy
Anticoagulation (AC) therapy is the mainstay pharmacologic treatment for the management and prevention of blood clots. Commonly referred to as "blood thinners," anticoagulants function to reduce the risk of morbidity and mortality associated with thrombotic events by helping to prevent and/or dissolve existing blood clots, as well as slow down the body's process of generating new blood clots (Witt et al., 2018).
Safeguarding Patients from Adverse Events
While AC therapy can be lifesaving, it can also pose serious risks for life-threatening bleeding events. The most common indicator for determining a patient's risk for bleeding is through laboratory tests that measure the blood's clotting capacity. The most common tests include the prothrombin time (PT) and the international normalized ratio (INR). The PT measures in seconds the amount of time it takes for the plasma portion of the blood to clot and the INR is a standardized number that is calculated based on the PT result. One of the most common reasons for performing PT/INR testing is to monitor the therapeutic levels of a common AC agent known as warfarin (Coumadin). Higher than normal PT/INR levels indicate a higher risk for bleeding events due to the body's impaired ability to clot blood. While on warfarin (Coumadin), it is essential that the PT/INR does not exceed therapeutic thresholds; otherwise, the risk for bleeding heightens (American Association for Clinical Chemistry [AACC], 2019a). Nurses often serve central roles in monitoring patients on warfarin (Coumadin) therapy through AC clinics or home-based monitoring. In the US, there are more than 3,000 AC clinics that serve more than 1 million patients. In collaboration with clinicians, licensed providers, or pharmacists, nurses working in these settings receive specialized training and are tasked with monitoring INR levels and communicating results. Many clinics now operate on evidence-based guidelines and algorithms to facilitate appropriate warfarin (Coumadin) dosing adjustments and appropriate follow up intervals. While AC clinics have historically been geared toward warfarin (Coumadin) monitoring, the surge in new oral AC agents has led to expanded AC clinics. The primary goals of expanded AC clinics are to mitigate bleeding risk or other AC-related complications, safeguard patient care, increase adherence to therapy, enhance patient knowledge about managing their therapy at home, and improve outcomes. Nurses are responsible for ensuring that all information about a patient’s lab results, dose changes, and responses to therapy are regularly communicated to the patient’s treating clinician, as well as the patient or caregiver (Barnes et al., 2016).
Key Teaching Points for Patients on AC
Nurses are responsible for ensuring that all patients and caregivers are adequately educated on the risks of bleeding while taking anticoagulants, as well as the risks associated with noncompliance. Studies demonstrate that effective medication counseling empowers patients to be active partners in their care, leading to better outcomes, such as enhanced treatment adherence, reduced adverse drug reactions, and lower overall healthcare costs (Hawes, 2018). Patients should be provided with verbal and printed information regarding the major complications of AC therapy and the importance of reporting side effects or adverse effects. The major complications include blood clotting due to under-dosing (or subtherapeutic dosing) or bleeding due to excessive thinning of the blood. The most serious bleeding events while on AC therapy include gastrointestinal and intracerebral bleeds, although bleeding can occur from any site. Patients and caregivers should be counseled on the importance of reporting all falls, accidents, or trauma. Patients should be advised to monitor for signs of abnormal bleeding and to seek medical attention for any of the following:
- Fall or head injury;
- Headache that is more severe than usual, confusion, weakness, or numbness, which can be signs of signal intracerebral bleeding;
- Hemoptysis;
- Hematemesis;
- Bleeding that will not stop (American Heart Association [AHA], 2016; Witt et al., 2018).
Nurses should also educate patients on specific lifestyle changes to reduce bleeding risk and promote safety while on anticoagulation therapy, such as:
- Wearing a medical alert bracelet;
- Shaving with an electric razor;
- Using a soft-bristle toothbrush and waxed floss;
- Avoiding activities with the potential for falling or self-injury (i.e. contact sports),
- Avoiding excessive alcohol consumption;
- Speaking with their clinician prior to starting medications that pose additional bleeding risk, such as non-steroidal anti-inflammatory drugs (NSAIDs, such as ibuprofen [Motrin, Advil] and acetylsalicylic acid [Aspirin]);
- Removing throw rugs that may increase fall risk;
- Wearing a helmet when bike riding (Haugen, 2019).
Type of Anticoagulation Therapy
The choice of AC therapy is based on several factors, including the severity of thrombosis or thrombophilia disorder, patient and clinician preference, adherence to therapy, and potential drug and dietary interactions. The 2018 ASH guidelines do not specify which medication is preferred for VTE treatment, but instead, guide the use of the different groups of medications and their clinical considerations. While nurses are not responsible for selecting the type of AC therapy, they should have a basic understanding of each medication, key drug interactions, reversal of each AC agent, if available, and management of acute bleeding events (Witt et al., 2018).
Vitamin K Antagonists (VKA)
VKAs are the oldest class of oral AC and have a variety of medical indications that extend beyond the treatment and prevention of VTEs, such as AFib, ischemic stroke, acute MI, patients with mechanical prosthetic cardiac valves, and patients undergoing orthopedic surgery. VKAs are commonly used as prophylaxis for VTE in high-risk patient populations, such as those with embolic peripheral and arterial disease. VKAs prevent coagulation by suppressing the synthesis of vitamin K-dependent factors. In other words, they act as antagonists to vitamin K, impairing the liver's ability to process vitamin K into clotting factors, thereby curbing blood clotting. The goal of VKA therapy is to decrease the clotting tendency of blood, but not to prevent clotting entirely. There are several types of VKAs, such as warfarin (Coumadin), acenocoumarol (Ascumar), phenprocoumon (Marcoumar), and coumatetralyl (Racumin). However, warfarin (Coumadin) is the most well-known and widely used oral AC in the US and will be the focus of discussion within this section (Tritschler et al., 2018).
As described above, patients on warfarin (Coumadin) require ongoing monitoring of INR to ensure therapeutic levels of the drug, which can fluctuate based on dietary intake. Since Vitamin K is found in many foods such as green leafy vegetables and certain oils, if the consumption of vitamin K-rich foods increases, the patient will require higher warfarin (Coumadin) dose to maintain therapeutic levels. Similarly, the contrary is also true; with reduced intake of foods rich in vitamin K, the warfarin (Coumadin) dose may need to be reduced to prevent bleeding. If patients skip a dose (or take an extra dose) of warfarin (Coumadin), the warfarin (Coumadin) level may be altered for several days, as both vitamin K and warfarin (Coumadin) levels rise gradually. Further, various medications, dietary supplements, and herbal preparations can interfere with the metabolism of warfarin (Coumadin), which can lead to increased risk for bleeding or reduced efficacy of the medication, thereby increasing the risk for blood clot formation.
To reduce the risk for adverse bleeding events, patient education is critical. Nurses must educate patients on the importance of maintaining a consistent amount of vitamin K intake weekly and reporting any new medications or dietary preparations to prevent harmful interactions. Nurses should be knowledgeable regarding the most common warfarin (Coumadin) interactions, which include the following:
- Common drug interactions:
- Acetylsalicylic acid (Aspirin);
- NSAIDs, such as ibuprofen (Motrin)
- Acetaminophen (Tylenol) ;
- Antacids;
- Laxatives;
- Numerous types of antibiotics;
- Antifungal medications, such as fluconazole (Diflucan);
- Antiarrhythmic medications, such as amiodarone (Pacerone).
- Common herbal preparations and dietary supplement interactions:
- Coenzyme Q10 (ubiquinone),
- Garlic,
- Ginkgo biloba,
- Ginseng,
- Green tea,
- St. John's wort,
- Vitamin E.
- Aside from vitamin K, other foods and drinks that can potentially interact with warfarin (Coumadin) include the following:
- Cranberries or cranberry juice,
- Grapefruit or grapefruit juice,
- Alcohol,
- Black licorice (Mayo Clinic, 2020).
The target INR for patients with VTE on warfarin (Coumadin) is usually between 2 and 3. An unsafe/highly elevated INR (> 4.5) may lead to dangerous bleeding events. The only US Food & Drug Administration (FDA) approved reversal agent for VKA therapy is 4-factor prothrombin complex concentrate (PCCS) [Kcentra] with phytonadione (vitamin K1). PCCS is only used in the event of life-threatening bleeding while on VKA agent, is administered by slow intravenous (IV) injection, and Vitamin K should be administered concurrently to maintain therapeutic factor levels once the effects of PCCS (Kcentra) have diminished (FDA, 2018).
Direct Oral Anticoagulants (DOAC)
DOACs are a relatively novel group of AC that work by directly inhibiting specific proteins within the clotting cascade and include direct thrombin inhibitors and factor Xa inhibitors (American Society of Health-System Pharmacists [ASHP], n.d.). Patients on direct factor Xa inhibitors do not require monitoring to ensure therapeutic drug levels are maintained. Nurses should be well versed on the most common adverse effects of these agents which include nausea, dyspepsia, diarrhea, abdominal pain, anemia, and hemorrhage. Less commonly, patients can develop gastrointestinal ulcers, gastroesophageal reflux disease (GERD), acute kidney injury, esophagitis, and thrombocytopenia. The most common DOACs are listed below (Witt et al., 2018).
- Dabigatran (Pradaxa) is a direct thrombin inhibitor that exerts its AC effects by binding directly to thrombin, inhibiting both soluble and fibrin-bound thrombin. Since thrombin enables the conversion of fibrinogen into fibrin during the coagulation cascade, its inhibition prevents thrombus development.
- Rivaroxaban (Xarelto), apixaban (Eliquis), and edoxaban (Savaysa) are oral direct factor Xa inhibitors, which work by selectively and reversibly blocking the activity of FXa to prevent clot formation. While these agents affect FXa presence within the bloodstream and within a preexisting clot, they do not interfere with platelet aggregation (Tritschler et al., 2018).
Unfractionated Heparin (UFH)
Unfractionated Heparin (UFH) is a rapidly acting AC agent that collaborates with antithrombin to block clot formation. UFH binds to antithrombin and enhances its ability to inhibit FXa and FIIa quickly. While UFH does not break down blood clots, it functions to prevent new ones from forming, allowing the body to dissolve any existing thrombi gradually. UFH is administered via subcutaneous injection or as a continuous IV infusion. Dosing is based on body weight, and patients require frequent monitoring while on treatment with UFH to ensure appropriate dosing and safety. The primary advantages of UFH include its rapid onset and its relatively rapid termination following discontinuation of the medication. It is the preferred treatment for patients at heightened risk for bleeding due to its short half-life and reversibility. While the definitive half-life of UFH depends on the dose, the average half-life is about 30 to 90 minutes in most healthy adults (Longo, 2019). Nurses should be aware of protamine sulfate (Prosulf), which is commonly used for the reversal of UFH. Protamine sulfate (Prosulf) is administered as a slow IV injection and patients should be monitored closely due to the potential for severe hypotension and bradycardia. Resuscitation equipment should be available, and nurses should be prepared to treat the patient for shock as well as hemorrhage. Symptoms of urticaria, angioedema, pulmonary edema, anaphylaxis, and dyspnea have been reported. Nurses should monitor patients carefully for bleeding, which can occur up to nine hours later due to rebound of the effects of UFH. The patient’s blood pressure and pulse should be monitored every 15 to 30 minutes for at least two hours following administration. Further, nurses should expect to monitor coagulation tests, which are usually performed 5 to 15 min following administration of protamine sulfate (Prosulf), and then again in two to eight hours, if indicated (F.A. Davis Company, 2015b). Aside from uncontrolled bleeding events, potential side effects of UFH include redness and irritation at the injection site, elevations in liver enzymes, reduced bone mineral density, and HIT. HIT is a potentially life-threatening disorder that occurs in response to the administration of UFH and will be described in greater detail within the section on blood clotting disorders (National Blood Clot Alliance [NBCA], n.d.b.).
Low Molecular Weight Heparin (LMWH)
LMWHs are subcutaneous injections that work by inhibiting thrombin and factor Xa. Some of the most common LMWH agents include dalteparin (Fragmin), enoxaparin (Lovenox), nadroparin (Fraxiparin), and tinzaparin (Innohep). LMWHs are generally preferred over UFH or DOACs for the initial treatment of acute VTE secondary to their once-daily dosing regimen and reduced rate of complications. They carry a lower risk of HIT than UFH, and are less likely to be associated with reduced bone mineral density. The ASH advises against the routine monitoring of anti-Xa levels to guide dosing as monitoring demonstrates no difference in recurrent VTE or bleeding risk. Currently, anti-Xa tests are unreliable, and there is no established therapeutic range for LMWH. Protamine (Prosulf) may also be used as the antidote for patients with significant bleeding events on LMWH, although this is rarely required (Witt et al., 2018). Nurses usually serve essential roles with regards to teaching patients how to self-inject these agents. It is important that nurses advise patients not to eject the air bubble in the prefilled syringe prior to the medication; following the injection, patients should not aspirate or massage the site (F.A. Davis Company, 2015a).
Disorders of Excessive Blood Clotting
Excessive blood clotting disorders, also referred to as hypercoagulable conditions or thrombophilias, are conditions characterized by an increased propensity or predisposition to develop blood clots. These conditions may be inherited (genetic) or acquired and can affect both the venous and arterial circulation systems, however the majority affect the venous system, increasing the risk for VTEs (AACC, 2019a).
Inheritance Patterns of Disease
To understand the inherited disorders and appropriately counsel patients, it is essential that nurses first establish a basic understanding of inheritance patterns. Heterozygous is the term used to describe patients who have a mutation in only one copy of the gene, whereas homozygous describes a patient who has inherited a mutation in both copies of the gene (The US National Library of Medicine [NLM], 2020d).
Autosomal Dominant (AD)
In conditions that are AD, the abnormal gene is dominant and is located on one of the chromosomes. The term 'dominant' means that the patient only needs one mutated copy of the gene to be affected by the disorder. In some cases, an affected person inherits the condition from an affected parent. In other cases, the condition may develop from a new mutation in the gene and occur randomly in people with no history of the disorder in their family. As demonstrated in Figure 14 (left side), a person with an AD disorder has a 50% chance of having an affected child with one abnormal gene, and a 50% chance of having an unaffected child with two normal genes (NLM, 2020d).
Autosomal Recessive (AR)
In conditions that are AR, one abnormal gene must be inherited from each parent, as the patient requires two copies of the mutation to be affected by the disease. A carrier of the condition has one abnormal gene (recessive) and one normal gene (dominant) but is unaffected by the disorder. Therefore, as demonstrated in Figure 14 (right side), two carriers have a 25% chance of having an unaffected child with two normal genes, a 50% chance of having a child who is an unaffected carrier, and a 25% chance of having a child with two recessive genes affected by the disorder. AR disorders are not usually seen in every generation of an affected family (NLM, 2020d).
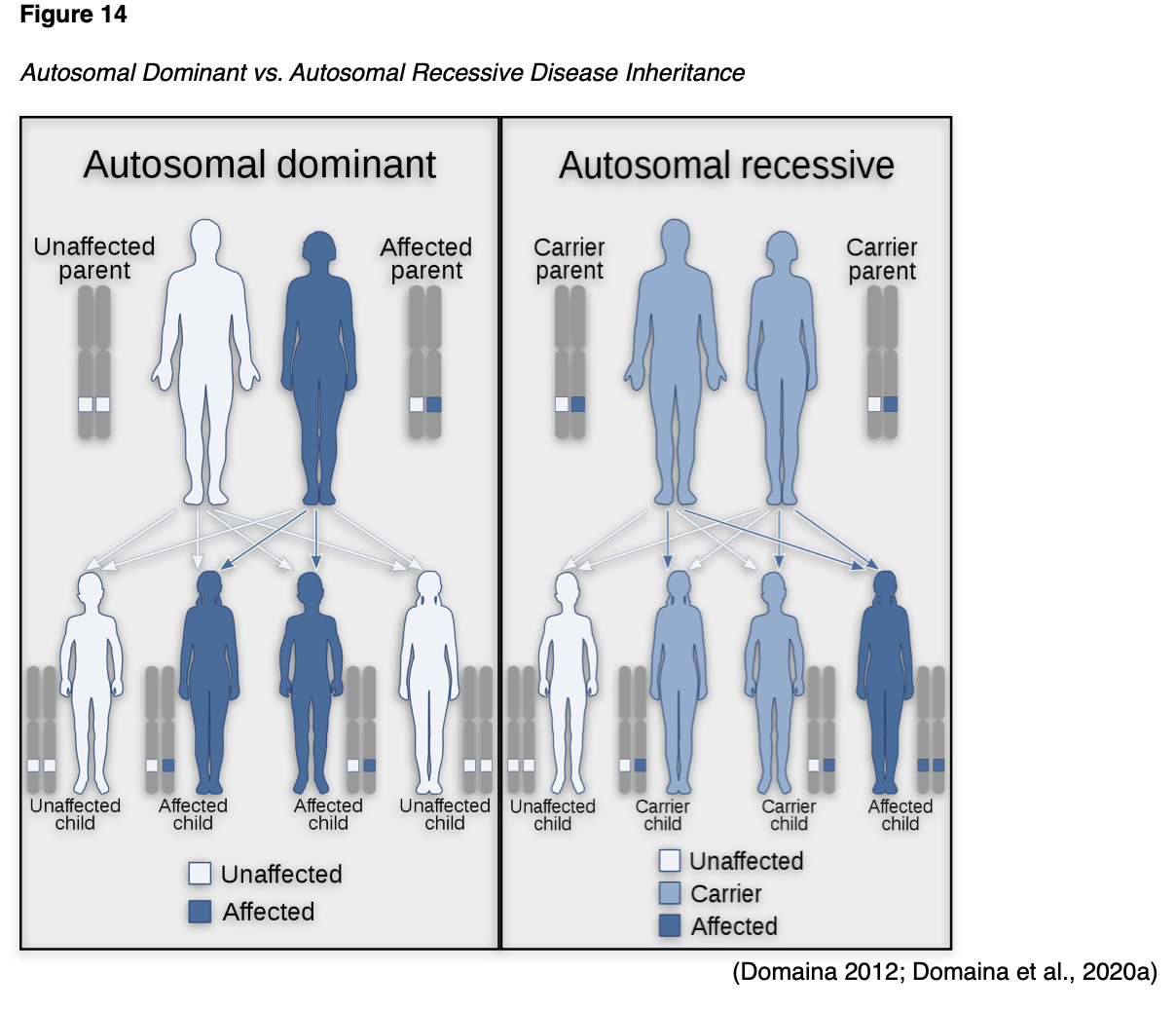
X-linked Dominant
X-linked dominant disorders are caused by mutations in genes on the X chromosome, one of the two sex chromosomes in each cell. Females have two X chromosomes, but a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. Since males only have one X chromosome, a mutation in the only copy of each cell's gene causes the disorder. While only a single copy of the mutation is enough to cause the disease in both males and females, males generally experience more severe symptoms than females. A characteristic trait of X-linked inheritance is that there is no male-to-male transmission; fathers cannot pass the X-linked traits to their sons since fathers only pass on their Y chromosome to their sons and their X chromosome to their daughters. As demonstrated in Figure 15, the sons of a male with an X-linked dominant disorder will not be affected, whereas 100% of his daughters will inherit the condition. The mother passes one of her X chromosomes to each child. Therefore, a woman with an X-linked dominant disorder has a 50% chance of having an affected daughter or son with each pregnancy (NLM, 2020d).
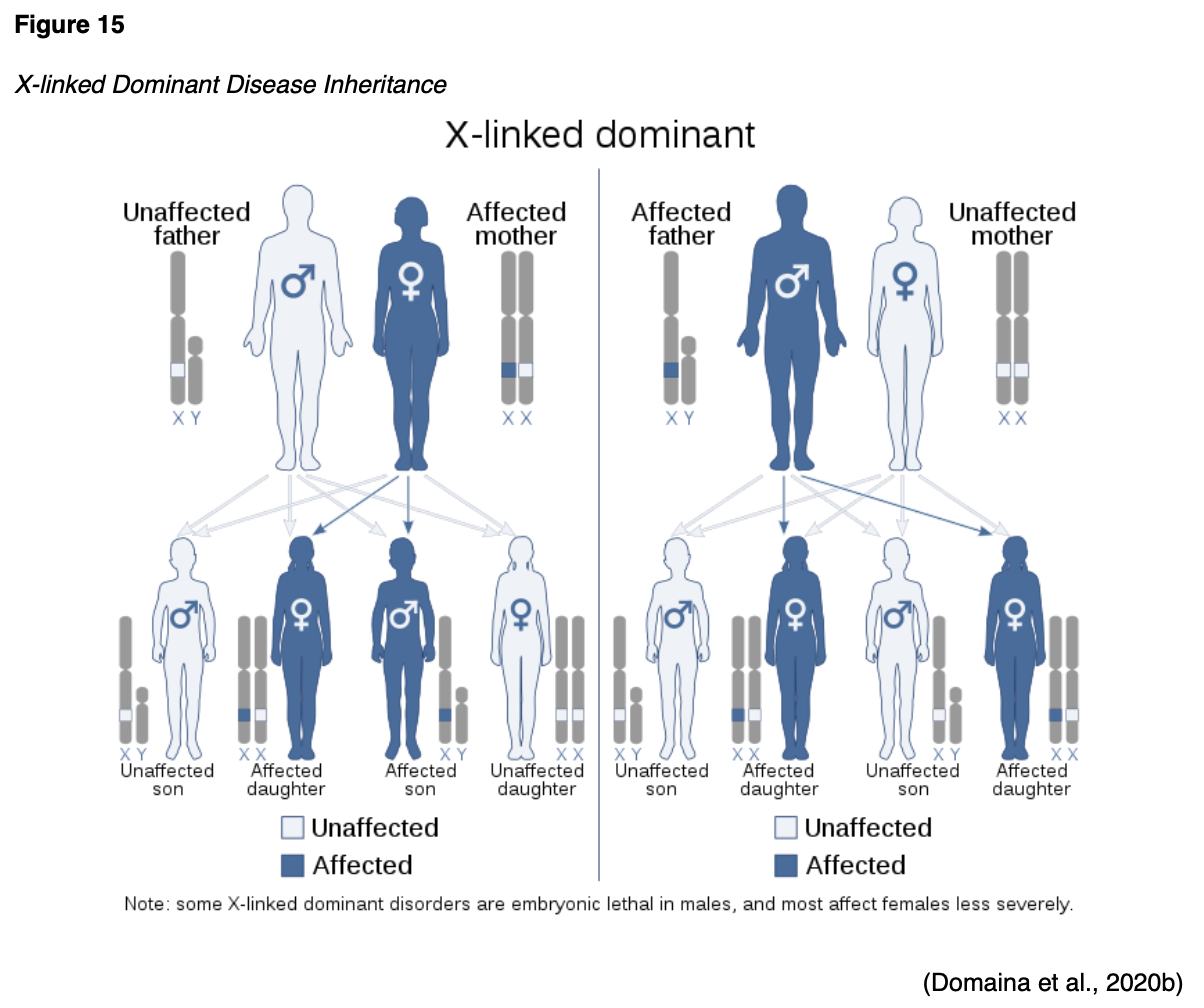
X-linked Recessive
X-linked recessive disorders are also caused by mutations in genes on the X chromosome. In males, one altered copy of the gene in each cell is sufficient to cause the condition; in females, a mutation would have to occur in both copies of the gene to cause the disorder. Since it is unlikely that females will acquire two altered copies of the gene, males are much more likely to be affected by X-linked recessive disorders than females. As demonstrated in Figure 16, the sons of a man with an X-linked recessive disorder will not be affected, while all of his daughters will carry one copy of the mutated gene. A woman with an X-linked recessive disorder has a 50% chance of having sons affected by the disorder, and a 50% chance of having daughters who carry one copy of the mutated gene (NLM, 2020d).
Since patients with thrombophilia disorders are already at heightened risk for blood clotting, nurses should counsel patients on ways to mitigate their risk by controlling modifiable risk factors, such as oral contraceptive use, hormone replacement therapy, and smoking. Other factors that increase the risk of blood clots include increasing age, obesity, trauma, surgery, and pregnancy (NLM, 2020d).
Factor V Leiden Thrombophilia (FVL)
A mutation in the FV gene causes FVL. FVL gene mutations inactivate factor V more slowly than normal, allowing the clotting process to remain active longer than usual. As a result, this increases the risk for VTE (Genetic and Rare Diseases Information Center [GARD], 2017a). Considered the most common inherited form of thrombophilia, FVL affects approximately 3-8% of people with European ancestry. Although the condition increases the risk of VTE, only about 10% of affected individuals ever develop abnormal clots, with DVT and PE among the most common. FVL is inherited in an autosomal dominant manner. The chance of developing abnormal blood clots depends on whether the individual has inherited one or two copies of the FVL mutation in each cell. Those who inherit two copies of the mutation (one from each parent) are at higher risk of developing a thrombus than those who inherit one copy of the mutation. The majority of individuals affected by the condition have one "normal" F5 gene and one with the factor V Leiden gene mutation. In the general population, 1 in 1,000 people per year (PPY) develop an abnormal blood clot. In patients with one copy of the FVL mutation, the risk of thrombus formation is 3 to 8 in 1,000 PPY, whereas two copies of the mutation heighten the risk to 80 in 1,000 PPY (NLM, 2020a).
Prothrombin G20210A Mutation (Prothrombin Thrombophilia)
Prothrombin G20210A gene mutation is also referred to as prothrombin thrombophilia. Patients with this mutation produce excess amounts of prothrombin (FII), which leads to an abundance of thrombin in the circulation, thereby increasing the tendency to form VTE. Considered the second most common inherited form of thrombophilia, G20210A mutation is present in about 1 in 50 people in the Caucasian population and is more common in those of European ancestry. Prothrombin thrombophilia follows autosomal dominant inheritance, and the risk of developing a VTE is linked to the inheritance pattern of the condition. Heterozygous patients have a risk of 2 to 3 in 1,000 PPY, whereas those who are homozygous have risk of up to 20 in 1,000 PPY (NLM, 2020c). Management of this condition depends on whether a blood clot has occurred and if there are additional risk factors. Asymptomatic patients who have never had a VTE are not usually treated with routine AC therapy (GARD, 2017b). AC therapy is reserved for patients who have developed a VTE or are considered to be at risk of developing another VTE (NLM, 2020c).
Deficiencies of Natural Proteins that Prevent Clotting
Antithrombin (AT) deficiency. Considered a rare condition affecting males and females in equal proportions, AT deficiency affects 1 in 3,000 to 1 in 5,000 individuals within the US. Only about 1% of patients who develop a VTE or PE have congenital AT deficiency. Caused by mutations in the SERPINC1 gene, patients with inherited deficiencies in AT have reduced levels of AT circulating within their blood. As previously mentioned, AT functions to limit blood clotting, serving as a natural anticoagulant. As the primary inhibitor of thrombin, those affected by AT deficiency are at a lifelong predisposition to developing VTEs. The majority of patients affected by inherited AT develop their first VTE before age 40 and usually manifest as a DVT or SVT, as arterial thrombosis is very rare in AT deficiency. About 40% of patients with AT deficiency develop a PE, which makes the condition particularly dangerous. AT deficiency follows an autosomal dominant inheritance pattern, but unlike other conditions previously discussed, this one has variable clinical penetrance in heterozygotes. This means that not all patients who inherit one copy of the altered genetic mutation will be affected. Some may never endure a VTE event. Unfortunately, homozygotes with two copies of the altered gene are primarily seen in newborns who do not survive. Of note, AT deficiency is not always inherited. It can be acquired infrequently as a complication of another medical disorder, such as surgery, trauma, pregnancy, childbirth, metastatic cancer, and liver failure. This is important because while inherited AT deficiency increases the risk for blood clots, acquired AT deficiency usually does not (NBCA, n.d.a).
Deficiency in Protein C/S. Since protein C and protein S function as part of the body's innate anticoagulation mechanisms, if there is a deficiency in one or both of these proteins, or if either protein's functioning is impaired, clot formation can occur unregulated, leading to hypercoagulable states. Patients with deficiencies in either or both proteins are at increased risk for VTE events. These conditions are usually inherited, but can also be acquired (NLM, 2020b).
Protein C Deficiency. Inherited protein C deficiency is caused by a genetic mutation in the PROC gene. The degree of deficiency can be mild to severe. In milder forms of the condition, the patient may never endure a blood clot. Concerning inheritance patterns, protein C deficiency is primarily inherited in an autosomal dominant pattern. There are two types of inherited protein C deficiencies, ranging from mild to severe. The severity of the deficiency is determined by the number of PROC gene mutations the patient has. In the rare instance that a patient inherits two mutated copies of the protein C gene (one from each parent), the disease can be very severe, heightening the risk for intracranial thromboembolism in infants and VTE during childhood (NLM, 2020b).
Protein S Deficiency. Inherited protein S deficiency is caused by a genetic mutation in the PROC1 gene and is primarily inherited in an autosomal dominant manner. Patients who inherit two mutation copies are at higher risk for developing a more severe form of thrombosis called purpura fulminans, which is a life-threatening condition involving severe clotting throughout the body. Purpura fulminans is characterized by blood spots, bruising, and discoloration of the skin from coagulation in the small blood vessels within the skin, which can lead to tissue necrosis (AACC, 2019b). There is only one FDA-approved treatment for the prevention and treatment of VTE and purpura fulminans secondary to severe congenital protein C deficiency. Protein C concentrate [Human] (Ceprotin) is an IV injection generated from human plasma that was first approved for use in 2007. Since it is human plasma, it carries a risk of transmission of infectious agents, particularly viruses, as well as bleeding events. The dose, administration frequency, and duration of treatment are dependent upon the severity of the protein C deficiency and the patient's age and plasma level of protein C. High doses of IV protein C concentrate (Ceprotin) help thin the blood and protect from blood clots. It can also be used as a preventative treatment against blood clots during surgery, pregnancy/delivery, prolonged immobility, or sepsis. However, it is not routinely used in clinical practice. In accordance with FDA guidelines, treatment can only be administered under the direct supervision of a hematology specialist experienced in replacement therapy with coagulation factors and inhibitors, and in a setting where monitoring of protein C activity is possible (FDA, 2007).
Hyperhomocysteinemia
Homocysteine is a type of amino acid, which is a chemical the body uses to generate proteins. Under physiologic conditions, vitamin B6, vitamin B12, and folic acid break down homocysteine in the blood and transform it into substances the body needs, leaving minimal homocysteine circulating within the bloodstream. Hyperhomocysteinemia is characterized by increased levels of homocysteine in the blood, associated with a marked increase in the risk for VTE. Elevated homocysteine levels carry prothrombotic properties, causing vascular injury, platelet accumulation, and increased risk for forming occlusive thrombosis. Approximately 10% of primary episodes of VTE are due to elevated homocysteine levels (MedlinePlus, 2018a).
Hyperhomocysteinemia can be inherited or acquired. It is generally ignited by homocystinuria, a rare inherited disorder that prevents the body from breaking down certain proteins (methionine), leading to an abnormal accumulation of homocysteine and its metabolites in blood and urine. Homocystinuria is inherited in an autosomal recessive pattern. The accumulation of homocysteine and its metabolites is caused by disruption of any of the three interrelated pathways of methionine metabolism—deficiency in the cystathionine B-synthase (CBS) enzyme, defective methylcobalamin synthesis, or abnormality in methylenetetrahydrofolate reductase (MTHFR). Patients who inherit the condition usually have a mutation in MTHFR, causing increased homocysteine production (Mandava, 2018). Aside from VTE events, symptoms can vary and may include fatigue, numbness, tingling, weight loss, as well as cognitive impairment and cognitive decline. Treatment remains controversial and generally involves homocysteine-lowering treatments such as pyridoxine (Vitamin B6), folic acid (Vitamin B9), and hydroxocobalamine (Vitamin B12a). Several studies have demonstrated the benefit of using folate supplementation to lower homocysteine levels, thereby reducing cardiovascular and VTE risk (Son & Lewis, 2020).
Antiphospholipid Antibody Syndrome (APS)
APS is an acquired autoimmune condition in which the body’s own antibodies attack and damage phospholipids. As previously described, phospholipids are present on the lining of blood vessels and play an important role in maintaining the integrity of cell wall membranes. When the antibodies attack phospholipids, the blood cells become damaged, degrade, and lead to thrombosis formation in both the veins and arteries. Thrombosis is the hallmark of the condition, with VTEs more common than arterial thromboses. In APS, excessive and unregulated blood clot formation can block the blood flow to organs, impacting multiple body systems, leading to stroke, MI, renal damage, as well as venous complications, including PE and DVT. The condition can be fatal in severe cases. To further add to the complexity of the condition, patients who have APS are at higher risk for thrombocytopenia, as the antibodies either destroy the platelets, or the platelets are used up during the clotting process; this heightens the risk for mild to serious bleeding events (NHLBI, n.d.a).
APS is an acquired disorder. While the definitive etiology is not completely understood, it is usually associated with infection, cancer, certain medications, and some rheumatologic conditions such as systemic lupus erythematosus (SLE). Approximately 10% of patients who have SLE also have APS, and approximately 50% of all patients with APS also have another autoimmune or rheumatic disorder. Although rare, some patients affected by APS develop catastrophic antiphospholipid syndrome (CAPS), which is a condition characterized by the development of an excessive number of blood clots within weeks or months. While APS has no cure, AC therapy can help prevent complications by inhibiting blood clot formation and preventing existing clots from getting larger (NHLBI, n.d.a).
Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH is a rare acquired and potentially life-threatening blood disorder characterized by hemolytic anemia (destruction of RBCs), bone marrow failure, and abnormal thrombosis formation (especially in the hepatic, portal, mesenteric, splenic, and renal veins), and in the brain (cerebral veins). Clinical manifestations include weakness, fatigue, pallor, shortness of breath, thrombosis, and less commonly, hemorrhage. Patients commonly display hemoglobinuria (abnormally high hemoglobin levels in the urine) manifested by dark or blood-colored urine. This finding is most prominent in the morning as the urine concentrates overnight during sleep. PNH affects between 1 and 5 per million people and is most common in adults aged 35 to 40. Up to 30% of newly diagnosed cases evolve from aplastic anemia, and up to 30% develop following immunosuppressive therapy for aplastic anemia (Johns Hopkins Medicine [JHM], n.d.). Further, up to 30% of those affected by PNH develop thrombosis, especially VTEs, although arterial blood clots can occur. While the median survival for the condition is about 10 years, some patients can survive for decades with only minor symptoms (National Organization for Rare Disorders [NORD], 2019b).
PNH is caused by a somatic mutation in the phosphatidylinositol glycan anchor biosynthesis class A (PIGA) gene. This is not an inherited condition and cannot be passed down to children of affected individuals. Instead, this is an acquired condition in which the gene changes occur throughout the course of a patient's lifetime and are only present in certain cells. In PNH, somatic mutations of the PIGA gene occurs within the hematopoietic stem cells, leading to the development of abnormal blood cells, which subsequently multiply. The proportion of abnormal blood cells is directly correlated with the severity of the condition. Chronic hemolysis contributes to the development of blood clots, which are most common in the abdominal veins, which can lead to Budd-Chiari syndrome, a rare condition caused by occlusion of the hepatic veins that drain the liver. Patients develop severe abdominal pain, ascites, jaundice, and hepatomegaly (NORD, 2019b).
Heparin-Induced Thrombocytopenia (HIT)
HIT is a potentially fatal acquired disorder that occurs in response to the administration of UFH. Affected patients form antibodies against heparin and the platelet factor-4 (PF4) complex. Immune complexes of HIT antibodies and PF4/heparin bind to the surface of platelets and activate them. Activated platelets adhere to the blood vessel lining, promote clotting activity, and clump together, causing an overuse of platelets, thereby inducing thrombocytopenia. Damage to the blood vessel wall and platelet clumping associated with HIT can lead to the formation of blood clots despite the presence of UFH. UFH administration is the most common cause of the condition. However, LMWH agents and fondaparinux (Arixtra) each also carry a reduced risk, respectively (Indiana Hemophilia & Thrombosis Center [IHTC], 2020). The prevalence of HIT can occur in up to 5% of patients receiving some form of heparin therapy, and if left untreated, 50-89% of affected patients will develop a thrombosis. While it remains unclear why some patients develop HIT while others do not, some of the strongest risk factors include the following:
- Longer duration of heparin therapy (longer than five days);
- Higher doses of heparin;
- The indication for treatment, with surgical (orthopedic) and trauma patients at highest risk, followed by patients with cardiovascular disease and cardiovascular interventions;
- Gender, with females affected more commonly than males (IHTC, 2020; Salter et al., 2016).
In most cases, UFH is administered for 7 to 10 days before the onset of HIT symptoms, and the clinical presentation can be variable. The most common clinical signs include bleeding, bruising, ecchymosis, new thrombosis formation, or an increase in the size of a current thrombosis despite being on anticoagulation therapy (IHTC, 2020). For most patients, the first sign is thrombocytopenia identified on routine CBC test, in which the platelet count declines by at least 50% from baseline. Therefore, even if the patient's platelet count remains within normal limits, a 50% reduction while receiving UFH or a LMWH should still warrant clinical suspicion for the condition (Salter et al., 2016). Nurses have a responsibility to monitor results and facilitate the early communication of any suspicious findings or abnormalities to the provider. Nurses should remember that the key to mitigating risks and complications associated with HIT is the immediate discontinuation of all forms of heparin. This includes heparin-coated catheters, as well as any LMWH agents. The platelet count may take up to 30 days to recover, and due to the increased risk for thrombosis formation, most patients will still require an alternative form of AC therapy (IHTC, 2020).
In HIT, nurses should understand that oral AC options are limited to non-heparin anticoagulant agents such as direct thrombin inhibitors (lepirudin [Refludan], argatroban [Acova], bivalirudin [Angiomax]), or danaparoid (Orgaran), an antithrombin-depending inhibitor of FXa. These are considered the drugs of choice for patients with HIT since they don't require antithrombin-III for their desired anticoagulation effects. It is important that warfarin (Coumadin) is not used in patients with HIT due to the increased risk for developing warfarin (Coumadin)-induced skin necrosis and venous limb gangrene. In patients where warfarin (Coumadin) may have already been initiated, the recommendation is to reverse the effect with vitamin K and replete protein C and S, which will help prevent gangrene of the limb.
Platelet transfusions are not routinely advised for the management of HIT; however, they can be considered in patients who are actively bleeding or undergoing invasive procedures that pose an additional bleeding risk. Nurses should educate patients on the need for continued AC treatment. The American College of Cardiology (ACC) recommends that all patients with HIT are treated with anticoagulation for at least four weeks, which may extend up to six months for patients affected by a thrombosis (Salter et al., 2016).
Disseminated Intravascular Coagulation (DIC)
DIC is a complex condition characterized by the systemic activation of coagulation, leading to widespread intravascular fibrin formation, organ failure, and/or profuse (internal and external) bleeding. DIC is always a secondary disorder that occurs in response to an underlying condition, causing the activation of the coagulation cascade, the release of cytokines, and overuse and increased consumption of the body's supply of platelets and clotting factors. In DIC, small blood clots form throughout the body's small and midsized vessels, obstructing blood supply to organs and contributing to organ failure. While DIC can be characterized by both clotting and bleeding, one or the other usually predominates. The condition is highly convoluted and can be attributed to a multitude of diseases or disorders, but most commonly develops as a result of severe infection such as sepsis, systemic inflammatory conditions, trauma, or as a byproduct of exposure of procoagulant materials within the bloodstream from malignancy or obstetric complications. The presentation of the condition is variable but is generally characterized by severe bleeding that occurs internally and externally. There are two forms of DIC; acute and chronic. Acute DIC develops over a few hours or days and causes serious bleeding. Chronic DIC develops over weeks to months and generally does not lead to excessive bleeding but is more closely associated with increased thrombosis. However, in both acute and chronic DIC, blood clots often form or travel to the lungs resulting in PE (Levi & Scully, 2018).
DIC treatment primarily focuses on identification, management, or elimination of the underlying condition. Interventions are primarily supportive and nursing care focuses on treating the hemodynamic instability of the condition, monitoring respiratory and cardiac status, and administering medications. Some patients will need adjunctive pharmacological therapy aimed at the coagulation system, either to control the bleeding or thrombosis. For instance, UFH may be used for chronic DIC when blood clotting is problematic, whereas excessive blood loss may require blood product transfusions. Antibiotics may be required when infection or sepsis is the underlying factor. The majority of the therapeutic options for DIC are not based on high levels of evidence and treatment is usually an algorithm-based approach to managing the underlying condition and clinical manifestations as they arise. Nurses should initiate bleeding precautions such as avoiding razors, using a soft toothbrush, limiting venipunctures, and minimizing the risks of bleeding from friction, injury, or pressure (Thachil, 2016).
Bleeding Disorders
When the physiologic mechanisms of blood clotting fail, are deficient, or insufficient, bleeding can ensue. Bleeding disorders are a group of conditions that are characterized by impaired hemostasis or the inability to properly form a blood clot. Parallel to excessive blood clotting disorders, bleeding disorders can also be inherited or acquired. However, acquired disorders are more common. Patients with genetic deficiencies experience life-long recurrent external and internal bleeding episodes that may occur spontaneously or in response to an injury (Longo, 2019). External bleeding can occur from dental procedures, in the oral or nasal cavities, or from superficial skin cuts that don't clot or stop bleeding. Internal bleeding can occur in the joints, muscles, soft tissues, and closed spaces, including intracranial hemorrhage. Women affected by bleeding disorders most commonly experience menorrhagia, or heavy menstrual bleeding and cramping (National Hemophilia Foundation [NHF], n.d.).
Hemophilia
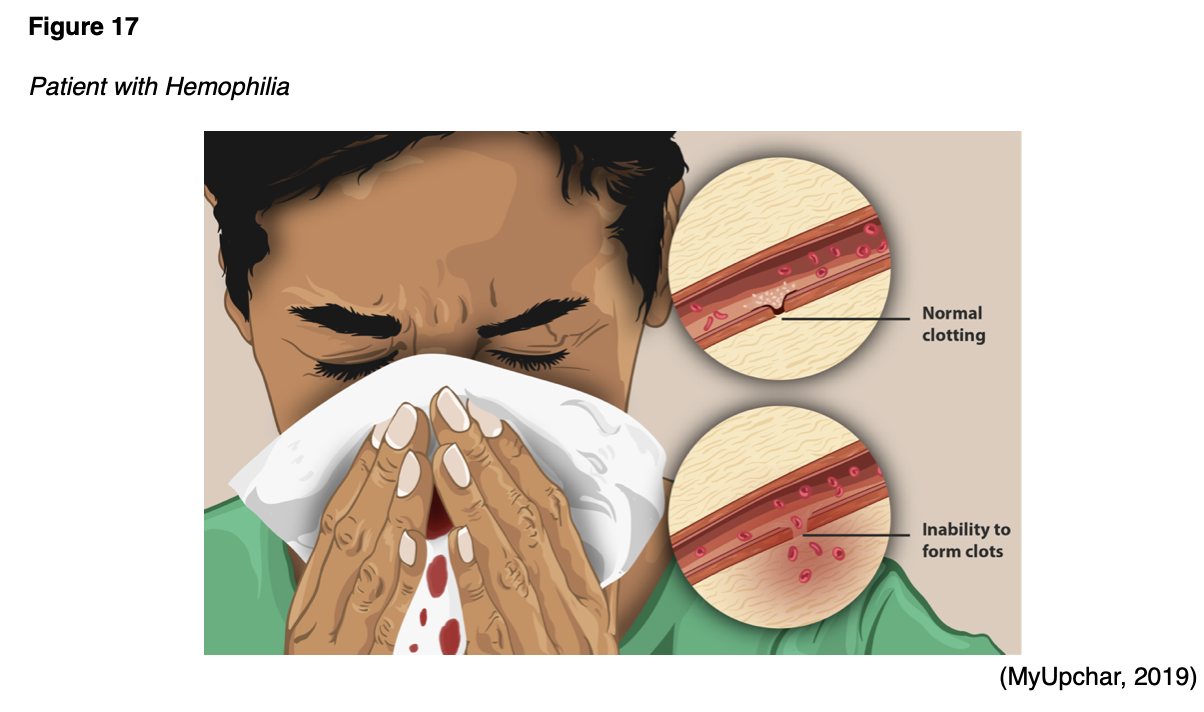
Hemophilia is a factor deficiency disorder, inherited in an X-linked recessive manner, and is comprised of several types, but the following two are the most common; hemophilia A (FVIII deficiency) and hemophilia B (FIX deficiency). According to the CDC (2020a), hemophilia A is four times more common than hemophilia B, affecting 1 in 5,000 male births, and about 400 babies annually. Within the US, the majority of patients with hemophilia are diagnosed during infancy, between 1 month and 36 months, depending on the severity of the condition. Two-thirds of the babies born with hemophilia have a known family history of the condition; about one-third have no known family history. As shown in Figure 17, patients with hemophilia bleed longer than other people, as they cannot properly form a blood clot. There is no cure for this lifelong condition, subject to periods of acute bleeding, prolonged bleeding, and bleeding crises. Since FVIII and FIX serve more important roles in deep tissues, hemophilia tends to cause more bleeding within the joints and muscles (CDC, 2020a). There are federally funded hemophilia treatment centers (HTCs) located throughout the US, which provide comprehensive medical care to patients suffering from this condition. Given the complexity of the condition and its life-threatening nature, specialized medical teams led by skilled hematologists are vital (NHF, n.d.).
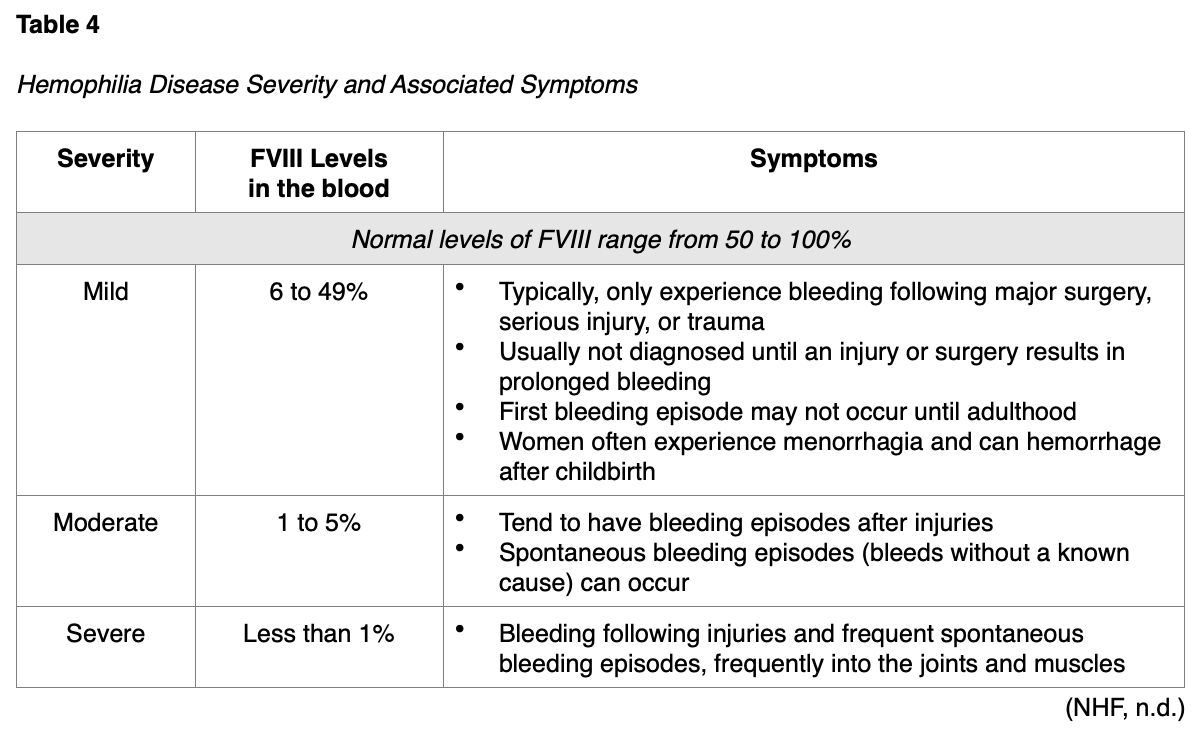
Hemophilia A also called classic hemophilia, is caused by a genetic mutation in FVIII, causing it to be defective or missing entirely. The condition is most commonly passed from parent to child. Hemophilia B, also called Christmas disease, is caused by a deficiency or absence of FIX. As shown in Table 4, the severity of hemophilia (for either A or B) exists on a scale ranging from mild to severe and the severity of symptoms is directly correlated with the stage (NHF, n.d.).
Treatment for hemophilia is centered on controlling bleeding by replenishing or replacing the missing blood clotting factor. In hemophilia A, the primary medication is concentrated FVIII product, referred to as the clotting factor. Recombinant factor products are created synthetically instead of through human-derived plasma products, which are safer. Currently, around 75% of patients with hemophilia take a recombinant FVIII product. The treatment for hemophilia B is essentially the same, except the primary medication is concentrated FIX product, which is also available as a recombinant factor product. These factor therapies are infused intravenously. Patients with severe hemophilia may receive treatments regularly, as prophylaxis, to ensure enough clotting factor is maintained within the bloodstream to prevent significant bleeding. Aminocaproic acid (Amicar) is an oral antifibrinolytic agent that may help prevent the breakdown of blood clots. It is usually given prior to dental procedures or to treat epistaxis. Nurses should understand that a dose of clotting factor is given first to help form a clot, which is then followed by aminocaproic acid (Amicar) to preserve the clot and keep it from being broken down prematurely. Desmopressin acetate (DDAVP) is a synthetic form of vasopressin, the body's natural antidiuretic hormone that helps stop bleeding. It is dispensed as both an injectable and a nasal spray and is used in patients with mild hemophilia for the management of joint and muscle bleeds, bleeding in the mucous membranes of the mouth or nose, or before and following surgery. Additional nursing care of the patient with hemophilia includes relieving pain, particularly joint pain, which can be achieved by immobilizing affected joints. Nurses should advise patients to elevate the affected area and to apply a cold compress to the active bleeding site to help with vasoconstriction and discomfort (NHF, n.d.).
About 15 to 20% of patients with hemophilia (or 1 in 5 with hemophilia A or 3 in 100 with hemophilia B) will develop an antibody or inhibitor. An inhibitor prevents the clotting factors from being able to clot the blood and stop the bleeding. Inhibitors make it significantly more challenging to control a bleeding episode because they counteract the treatment and destroy it. When this occurs, the management of bleeding episodes becomes increasingly complex, as the patient generally requires more and/or different types of clotting factors. The reasons why some patients develop inhibitors are not well understood. They most commonly develop during the first 50 infusions of clotting factor concentrates; however, they can potentially develop at any time. Patients who develop inhibitors tend to endure more joint disease and other complications from bleeding (CDC, 2019a).
von Willebrand Disease (VWD)
VWD is the most common bleeding disorder. It affects up to 1% of the US population; about 3.2 million (or 1 in every 100) people in the US are living with the disease. VWD is an incurable genetic disorder caused by defective or absent von Willebrand factor (vWF), thereby impairing platelet plug formation during the coagulation cascade and clotting process. It is carried on chromosome 12 and occurs equally in men and women; however, women are more likely to experience symptoms of increased bleeding during the menstrual cycle, pregnancy, and childbirth. There are three types of VWD, which can have different inheritance patterns, as outlined in Table 5. The majority of cases are type 1 and type 2, which are primarily inherited in an autosomal dominant pattern, whereas type 3 is predominantly inherited in an autosomal recessive pattern (CDC, 2019b).

vWF targets the skin and mucous membranes, including the lining of the nose, mouth, intestines, uterus, and vagina, so VWD tends to cause more bleeding. The main signs and symptoms of VWD include easy bruising, blood in stools, extended gum bleeding following dental procedures, and frequent episodes of bleeding that are difficult to stop. Epistaxis is the most common form of spontaneous bleeding in VWD, occurring more than five times per year and lasting for at least 10 minutes, requiring packing or cautery to stop the bleeding. Women commonly experience heavy menstrual bleeding with blood clots larger than the size of a quarter, usually soaking a pad every two hours (CDC, 2019b). Treatment is based on the severity of the condition, but since most patients have mild cases, treatment may only be required if the patient is undergoing surgery, a dental procedure, or after trauma from an accident or injury. The focus of pharmacological treatment is to control bleeding and prevent the breakdown of blood clots by increasing the release of vWF and FVIII into the bloodstream. This can be accomplished by administering vWF replacement therapy or synthetic recombinant FVIII agents. Nursing care is very similar to those with hemophilia. Desmopressin acetate (DDAVP) is often administered to patients with Type 1 and 2 VWD to facilitate the release of more vWF or FVIII. Antifibrinolytic therapy, such as Aminocaproic acid (Amicar), may also be used in conjunction with DDAVP or replacement therapy to help prevent the breakdown of blood clots (NHLBI, 2008). Patients with VWD Type 3 may also develop an inhibitor, which follows the same complex treatment trajectory as when it occurs in patients with hemophilia (CDC, 2019a).
Immune Thrombocytopenic Purpura (ITP)
ITP is an autoimmune bleeding disorder in which the immune system produces antibodies against platelets, destroying them and thereby preventing the blood from clotting normally. ITP affects both children and adults but is more common in children. In the US, ITP affects approximately 5.3 per 100,000 children per year and about 3.3 per 100,000 adults per year. According to NORD (2019a), among adults, the incidence of ITP increases with age and is more common in adults over the age of 60. Children often develop ITP after a viral infection, and they generally recover fully without treatment. Conversely, the disorder is more commonly chronic in adults and requires treatment. In adults, ITP can develop following viral infection, during pregnancy, from specific medications, or may present as part of an immune disorder. Patients often develop purpura or purple bruises on the skin or mucous membranes (including the mouth and nose) caused by bleeding from small blood vessels under the skin. They may also develop petechiae, which are pinpoint-sized red or purple dots on the skin that resemble a skin rash and most commonly occurs around the shin. Other signs or symptoms of ITP include excessive bruising, abnormally heavy menstruation, hematuria, and minor cuts and lacerations can take long periods to stop bleeding. In addition, ITP is commonly associated with debilitating fatigue and impaired quality of life for many affected patients (MedlinePlus, 2018b).
Among adults, first-line treatment for ITP is corticosteroids, such as prednisone (Deltasone), which suppresses the clearance of antibody-coated platelets and may also decrease the risk of bleeding by improving blood vessel lining cell function. Corticosteroid therapy is effective in increasing the platelet count in about 50% of patients, however extended corticosteroid use poses the risk for many side effects and important consideration. Nurses should ensure patients are properly educated on the risk of hyperglycemia, which can trigger the development of diabetes, osteoporosis, weight gain, acne, increased risk for infection, thinning of the skin, insomnia, and cataracts. The risks and benefits of long-term corticosteroid use must be carefully balanced (Mayo Clinic, 2019). In cases where the platelet count does not improve following corticosteroid therapy or if the patient develops severe bleeding, many patients will receive an IV immunoglobulin (IVIG) infusion in an outpatient setting every two to four weeks. IVIG is a blood product that comes from human plasma of healthy donors. When patients with ITP receive IVIG, it is thought to manipulate parts of the immune system that attack the platelets, thereby reducing the severity of the condition. Nurses must be prepared to respond to common adverse reactions to IVIG therapy such as infusion-related reactions, which can include fever, shaking, chills, hypotension, hives, rash, and fatigue. IVIG therapy carries a small risk that blood products may contain viruses that can cause infection; however, after the plasma is acquired from donors, it goes through an extensive purification process to mitigate the patient's risk of acquiring an infection. As a general rule, platelet transfusions are reserved for emergent situations because they are likely to be destroyed quickly by the autoantibodies. Nurses must educate patients with ITP on the importance of avoiding acetylsalicylic acid (Aspirin), NSAIDs such as ibuprofen (Motrin), warfarin (Coumadin), and anti-platelet medications such as clopidogrel (Plavix). These drugs interfere with platelet function and blood clotting, and can increase the risk for bleeding (MedlinePlus, 2018b; NORD, 2019a).
References
American Association for Clinical Chemistry. (2019a). Excessive clotting disorders. https://labtestsonline.org/conditions/excessive-clotting-disorders
American Association for Clinical Chemistry. (2019b). Protein C and protein S. https://labtestsonline.org/tests/protein-c-and-protein-s
American Heart Association. (2016). A patient’s guide to taking warfarin. https://www.heart.org/en/health-topics/arrhythmia/prevention--treatment-of-arrhythmia/a-patients-guide-to-taking-warfarin
American Society of Health-System Pharmacists. (n.d.). Managing and reversing direct oral anticoagulants: A discussion guide. Retrieved June 23, 2020, from https://ashpadvantagemedia.com/doacresources/files/doacresources-discussion-guide.pdf
Barnes, G. D., Nallamothu, B. K., Sales, A. E., & Froehlich, J. B. (2016). Reimaging anticoagulation clinics in the era of direct oral anticoagulants. Circulation: Cardiovascular Quality and Outcomes, 9(2),182-185. https://doi.org/10.1161/CIRCOUTCOMES.115.002366
Casas, A. L. F. (2019). Acute arterial embolism of the lower limb. https://www.intechopen.com/books/embolic-diseases-evolving-diagnostic-and-management-approaches/acute-arterial-embolism-of-the-lower-limb
The Centers for Disease Control and Prevention. (2019a). Inhibitors and hemophilia. https://www.cdc.gov/ncbddd/hemophilia/inhibitors.html
The Centers for Disease Control and Prevention. (2019b). What is von Willebrand disease? https://www.cdc.gov/ncbddd/vwd/facts.html
The Centers for Disease Control and Prevention. (2020a). Data & statistics on hemophilia. https://www.cdc.gov/ncbddd/hemophilia/data.html
The Centers for Disease Control and Prevention. (2020b). Stroke education materials for health professionals. https://www.cdc.gov/stroke/materials_for_professionals.htm
Domaina. (2012). Autosomal dominant [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:Autosomal_dominant_-_en.svg
Domaina, Kashmiri, & SUM1. (2020a). Autosomal recessive [image]. Wikimedia. https://en.wikipedia.org/wiki/File:Autosomal_recessive_-_en.svg
Domaina, Angelito7, & SUM1. (2020b). X-linked dominant disease inheritance [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:X-linked_dominant.svg
Domaina, Kashmiri, & SUM1. (2020c). X-linked recessive disease inheritance [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:X-linked_recessive_(2).svg
F.A. Davis Company. (2015a). Enoxaparin. https://davisplus.fadavis.com/3976/meddeck/pdf/enoxaparin.pdf
F.A. Davis Company. (2015b). Protamine sulfate. https://davisplus.fadavis.com/3976/meddeck/pdf/protaminesulfate.pdf
Garmo, C., Bajwa, T., & Burns, B. (2020). Physiology, clotting mechanism. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK507795/
Genetic and Rare Diseases Information Center. (2017a). Factor V Leiden thrombophilia. https://rarediseases.info.nih.gov/diseases/6403/factor-v-leiden-thrombophilia
Genetic and Rare Diseases Information Center. (2017b). Prothrombin-related thrombophilia https://rarediseases.info.nih.gov/diseases/10815/prothrombin-related-thrombophilia
Gross, P. L., Weil, P., & Rand, M. L. (2018). Harper’s illustrated biochemistry. (31st ed.). McGraw-Hill.
Haugen, N. (2019). Oral anticoagulants: Pharmacologic management update. https://www.myamericannurse.com/oral-anticoagulants-pharmacologic-update/
Hawes, E. M. (2018). Patient education on oral anticoagulation. Pharmacy (Basel), 6(2), 34-44. https://doi.org/10.3390/pharmacy6020034
Indiana Hemophilia & Thrombosis Center. (2020). Heparin-induced
thrombocytopenia. https://www.ihtc.org/heparin-induced-thrombocytopenia/
Jmarchn. (2014). Peripheral arterial disease [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:Blausen_Peripheral_Arterial_Disease_eng.svg
Joe D. (2007). Coagulation cascade with antithrombotic mechanisms [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:Coagulation_full.svg
Johns Hopkins Medicine. (n.d.). Paroxysmal nocturnal hemoglobinuria (PNH). Retrieved June 20, 2020, from https://www.hopkinsmedicine.org/kimmel_cancer_center/types_cancer/paroxysmal_nocturnal_hemoglobinuria_PNH.html
Leung, L. L. K. (2020). Overview of hemostasis. UpToDate. https://www.uptodate.com/contents/overview-of-hemostasis
Levi, M., & Scully, M. (2018). How I treat in brief: Managing disseminated intravascular coagulation. https://www.ashclinicalnews.org/education/managing-disseminated-intravascular-coagulation/?_ga=2.53709608.1489755650.1593259653-1187571219.1592763632
Lip, G., & Hull, R. D. (2020). Overview of the treatment of lower deep vein thrombosis (DVT). UpToDate. https://www.uptodate.com/contents/overview-of-the-treatment-of-lower-extremity-deep-vein-thrombosis-dvt
Longo, D. L. (2019). Harrison’s hematology and oncology. (3rd ed.). McGraw-Hill Education.
Mandava, P. (2018). Homocystinuria/homocyteinemia. https://emedicine.medscape.com/article/1952251-overview#a1
Mayo Clinic. (2019). Prednisone and other corticosteroids. https://www.mayoclinic.org/steroids/art-20045692
Mayo Clinic. (2020). Warfarin side effects: Watch for interactions. https://www.mayoclinic.org/diseases-conditions/deep-vein-thrombosis/in-depth/warfarin-side-effects/art-20047592
McCance, K. L., & Heuther, S. E. (2019). Pathophysiology: The biologic basis for disease in adults and children. (8th ed.). Elsevier.
MedlinePlus. (2018a). Homocysteine test. https://medlineplus.gov/lab-tests/homocysteine-test/
MedlinePlus. (2018b). Immune thrombocytopenia purpura (ITP). https://medlineplus.gov/ency/article/000535.htm
MyUpchar. (2019). Patient with hemophilia [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:A_woman_suffering_from_Hemophilia.png
National Blood Clot Alliance. (n.d.a). Antithrombin deficiency. Retrieved June 20, 2020, from https://www.stoptheclot.org/news/antithrombin-deficiency/
National Blood Clot Alliance. (n.d.b). Unfractionated heparin (UFH). Retrieved June 21, 2020, from https://www.stoptheclot.org/about-clots/blood-clot-treatment/unfractionated-heparin/#:~:text=2020%20NYC%20Marathon-,Unfractionated%20Heparin%20(UFH),body%2C%20to%20block%20clot%20formation.
The National Heart, Lung, and Blood Institute. (n.d.a.). Antiphospholipid antibody syndrome. Retrieved June 20, 2020, from https://www.nhlbi.nih.gov/health-topics/antiphospholipid-antibody-syndrome
The National Heart, Lung, and Blood Institute. (n.d.b). Stroke. Retrieved June 20, 2020, from https://www.nhlbi.nih.gov/health-topics/stroke
The National Heart, Lung, and Blood Institute. (2008). The diagnosis, evaluation, and management of von Willebrand disease. https://www.nhlbi.nih.gov/sites/default/files/media/docs/vwd.pdf
The National Heart, Lung, and Blood Institute. (2013a). Atrial fibrillation-induced
stroke [image].
https://upload.wikimedia.org/wikipedia/commons/3/34/Atrial_fib_stroke.jpg
The National Heart, Lung and Blood Institute. (2013b). Coronary thrombosis inducing a myocardial infarction [image]. https://commons.wikimedia.org/wiki/File:Heart_attack-NIH.gif
The National Heart Lung and Blood Institute. (2013c). Ischemic stroke [image].
https://upload.wikimedia.org/wikipedia/commons/4/44/Stroke_ischemic.jpg
National Hemophilia Foundation. (n.d.). Bleeding disorders. Retrieved June 23, 2020, from https://www.hemophilia.org/Bleeding-Disorders
National Organization for Rare Disorders. (2019a). Immune thrombocytopenia. https://rarediseases.org/rare-diseases/immune-thrombocytopenia/
National Organization for Rare Disorders. (2019b). Paroxysmal nocturnal hemoglobinuria. https://rarediseases.org/rare-diseases/paroxysmal-nocturnal-hemoglobinuria/
OpenStax College. (2013a). Platelet formation [image]. Wikimedia. https://en.m.wikipedia.org/wiki/File:1908_Platelet_Development.jpg
OpenStax College. (2013b). The steps of blood clotting and coagulation cascade [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:1909_Blood_Clotting.jpg
Persian Poet Gal. (2006). Blood clot (thrombus) diagram [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:Blood_clot_diagram.png
Rad, A., & Haggstrom, M. (2009). Hematopoiesis [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:Hematopoiesis_simple.svg
Salter, B. S., Weiner, M. M., Trinh, M. A., Heller, J., Evans, A. S., Adams, D. H., & Fischer, G. W. (2016). Heparin-induced thrombocytopenia: A comprehensive. clinical review. Journal of the America College of Cardiology, 67(21), 2519-2532. https://www.sciencedirect.com/science/article/pii/S0735109716324597?via%3Dihub
Son, P., & Lewis, L. (2020). Hyperhomocysteinemia. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK554408/
SportEX Journals. (2011). DVT [image]. https://www.flickr.com/photos/sportex/6634103129
Sved, A. (2019). Components of blood [image]. Wikimedia. https://commons.wikimedia.org/wiki/File:Components_of_blood.png
Thachil, J. (2016). Disseminated intravascular coagulation: A practical approach. Anesthesiology, 125, 230-236. https://anesthesiology.pubs.asahq.org/article.aspx?articleid=2516377
Tritschler, T., Kraaijpoel, N., Gal, G. L., & Wells, P. S. (2018). Venous thromboembolism: Advances in diagnosis and treatment. JAMA, 320(15), 1583–1594. https://doi.org/10.1001/jama.2018.14346
UNC Hemophilia and Thrombosis Center. (n.d.). What is a blood clot? Retrieved June 20, 2020, from https://www.med.unc.edu/htcenter/patient-care/clotting-disorders/blood-clot-education-1/what-is-a-blood-clot/
US Food & Drug Administration. (2007). Ceprotin [protein C concentrate (human)]. https://www.fda.gov/media/75033/download
US Food & Drug Administration. (2018). Kcentra (prothrombin complex concentrate, human). https://www.fda.gov/vaccines-blood-biologics/approved-blood-products/kcentra-prothrombin-complex-concentrate-human
The US National Library of Medicine. (2020a). Factor V Leiden thrombophilia. https://ghr.nlm.nih.gov/condition/factor-v-leiden-thrombophilia#definition
The US National Library of Medicine. (2020b). Protein C deficiency. https://ghr.nlm.nih.gov/condition/protein-c-deficiency#diagnosis
The US National Library of Medicine. (2020c). Prothrombin thrombophilia. https://ghr.nlm.nih.gov/condition/prothrombin-thrombophilia
The US National Library of Medicine. (2020d). What are the different ways in which a genetic condition can be inherited? https://ghr.nlm.nih.gov/primer/inheritance/inheritancepatterns
Witt, D. M., Nieuwlaat, R. Clark, N. P., Ansell, J., Holbrook, A., Skov, J., Shehab, N.,
Mock, J., Myers, T., Dentali, F., Crowther, M.A., Agarwal, A., Bhatt, M., Khatib, R., Riva, J. J., Zhang, Y., & Guyatt, G. (2018). American society of hematology 2018 guidelines for management of venous thromboembolism: Optimal management of anticoagulation therapy. Blood Advances, 2(22). 3257-3291. https://doi.org/10.1182/bloodadvances.2018024893