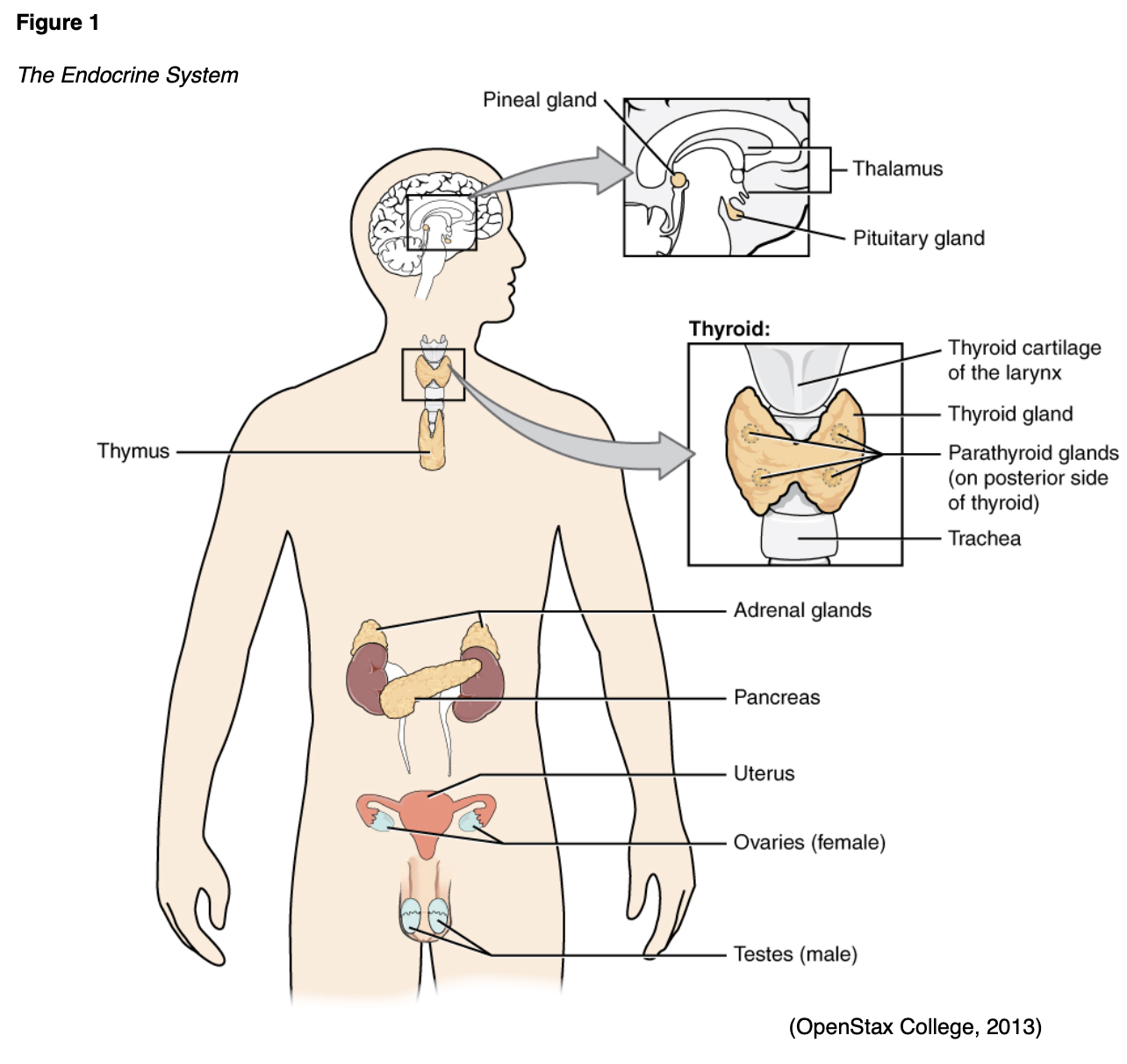
The purpose of this module is to provide the nurse with an overview of the pathophysiology, risk factors, clinical presentation, diagnosis, evidence-based management guidelines, and potential complications of primary endocrine disorders leading to hormonal deficiencies, including hypothalamus and pituitary disorders, growth hormone deficiency, DI, hypothyroidism, and hypogonadism.
...purchase below to continue the course
) or magnetic resonance imaging (MRI) can help visualize the hypothalamus, pituitary, and surrounding structures for the presence of a mass or lesion. Visual fields should be assessed if a pituitary mass is found to be the cause of hypopituitarism (Corenblum, 2020; Gouden & Jialal, 2020).
Treatment. Since hypopituitarism is a complex medical condition, it is best managed by an interprofessional team consisting of a neurosurgeon, an endocrinologist, a pathologist, a radiologist, a primary health care provider (HCP), and an ophthalmologist. Weight should be monitored closely, and a dietary consult should be considered for patients with anorexia or weight fluctuations. Parents of children with GH deficiency should be offered a referral to mental health counseling due to the associated stress for the patient and their parents (Fleseriu et al., 2016; Gounden & Jialal, 2020).
Treatment for hypopituitarism includes the replacement of the hormones that are secreted by the target glands. If left untreated, the loss of all anterior pituitary hormone function is fatal. Treatment should also focus on the management of the underlying cause, which may include the surgical removal of a tumor (Mayo Clinic, 2019b). As previously noted, since PRL deficiency is exceedingly rare, it is not routinely part of screenings. Clinical manifestations include an inability to lactate (Snyder, 2020a). There is currently no commercially available PRL replacement product in the US (Snyder, 2019c, 2020b).
The nurse should ensure patients with hypopituitarism receive adequate education that is inclusive of the following points:
- instruct the patient to wear a medical bracelet consistently identifying their disorder;
- emphasize the importance of life-long hormone replacement therapy and ensure the patient understands their medication regime, including prescription medications, herbals, and over-the-counter (OTC) drugs that may interact with their hormone therapies; and
- reinforce the importance of routine monitoring of hormone levels to maintain therapeutic levels and avoid excess or inadequate replacement (Gounden & Jialal, 2020).
Complications. Long-term complications include an increase in all of the following:
- mortality rates in those with nonfunctioning pituitary adenomas and deficiencies in ACTH or gonadotropin,
- the risk of cardiovascular disease due to decreased high-density lipoprotein (HDL) cholesterol levels and elevated low-density lipoprotein (LDL)/HDL ratio, and
- the incidence of cerebrovascular morbidity and mortality following pituitary radiotherapy (Corenblum, 2020; Willis, 2019).
The focus of this module will now shift to describe the presentation, diagnosis, and treatment of three common forms of hypopituitarism: GH deficiency, DI caused by ADH deficiency, and central/secondary hypothyroidism. We will then outline the various forms of hypogonadism, including secondary hypogonadism. Secondary adrenal insufficiency will be discussed in the previously mentioned course on adrenal dysfunction (Snyder, 2020a).
Growth Hormone Deficiency
GH (or somatotropin) is produced by somatotrophs in the anterior pituitary in response to the secretion of GH-releasing hormone (GHRH) by the hypothalamus. The production of GH by the pituitary is inhibited by the secretion of somatostatin (Chapman, 2019). The majority of GH is free/circulating, while approximately 40% is bound by GH-binding proteins (GHBP). It binds to GH receptors to facilitate growth and increased metabolism. The release of GH stimulates the production of insulin-like growth factor (IGF-1) by the liver, which thereby facilitates GH’s growth-promoting effects on the human skeleton (bone and cartilage tissue; El Sayed et al., 2020). GH deficiency presents differently in adults versus children (Snyder, 2020c). GH deficiency typically leads to a decreased lean-to-fat body mass, osteopenia, and in some cases, hyperlipidemia, cardiovascular disease, emotional changes (e.g., dysphoria, lethargy), and increased mortality in adults. In children, GH deficiency leads to short stature, as evidenced by decreased growth velocity and potentially delayed puberty (Corenblum, 2020; Snyder, 2020a).
Pathophysiology. The vast majority (75%) of cases of GH deficiency in adults result from a pituitary tumor or are secondary to the treatment of a pituitary tumor (Snyder, 2020c). Children with a deficiency in GH may present with proportionate dwarfism, in which the head, trunk, and limbs are small but proportionate to each other. The term dwarfism may be considered insensitive or offensive, and health care providers should consider using the term “short stature” instead, displaying abundant sensitivity to the emotional and psychological issues related to the condition (Mayo Clinic, 2018, 2019b).
Short stature secondary to hypopituitarism is often not recognized at birth; however, the early symptoms typically appear within the first months of life and are recognizable by six months, allowing early intervention. Children with short stature may appear overweight due to adipose tissue deposits in their lower trunk. They may have delayed secondary tooth eruption. The long-term outcome for an affected child will depend on the timing of disease presentation, diagnosis, and intervention for hypopituitarism. If the disease develops before puberty, secondary sex characteristics may be delayed or prevented (Willis, 2019).
Diagnosis. Adult patients at increased risk include those with known pituitary or hypothalamus disease, deficiencies in other pituitary hormones, or a history of GH deficiency (Snyder, 2020c). IGF-1 is the primary diagnostic laboratory test for GH deficiency. A bone age assessment is also recommended for young children with suspected GH deficiency. Bone age is typically calculated using a simple radiograph of the patient’s left hand or wrist (Richmond & Rogol, 2020a, 2020b). An IGF-1 level below normal in an adult patient with organic pituitary disease is diagnostic. Provocative testing for both children and adult patients with equivocal test results using arginine, clonidine, glucagon, or an insulin bolus can be considered for confirmation if required (Snyder, 2020c; Willis, 2019).
Treatment. In children, most experts recommend treatment with recombinant human GH, or somatropin (rhGH, Omnitrope, Norditropin, Humatrope, Genotropin), if the epiphyses are open (Rogol & Richmond, 2020). The long-term outcomes for children with GH deficiency depend on the timing of disease presentation, diagnosis, and intervention for hypopituitarism. If the disease develops before puberty, secondary sex characteristics may be delayed or prevented (Willis, 2019). Ideally, treatment may help patients achieve a normal height and increase their musculature (Cleveland Clinic, 2020).
The 2016 Endocrine Society guidelines regarding hypopituitarism in adults recommend the use of somatropin (Omnitrope) in most adults, although many experts question whether the limited clinical benefit of this treatment outweighs the high cost of this medication. Side effects of somatropin (Omnitrope) include peripheral edema, arthralgia, carpal tunnel syndrome, paresthesias, and glucose intolerance (Snyder, 2020c).
Diabetes Insipidus
DI is an uncommon disorder that results in fluid imbalances in the body, causing polyuria and polydipsia. It can be caused by an ADH deficiency related to a tumor, trauma, or other problem within the hypothalamus or pituitary gland. In addition, surgeries in the area of the pituitary, certain drugs such as glucocorticoids, or alcohol can result in DI (Hopper, 2015; Mayo Clinic, 2019a). There are multiple types of DI, as shown in Table 1 below.

The estimated prevalence of central DI is 1 per 25,000 people (Shahid et al., 2020). Risk factors for all types of DI consist of anything that damages the hypothalamus or pituitary, including surgery, head injury, infection, inflammation, increased intracranial pressure (ICP), or a tumor (Mayo Clinic, 2019a; NIDDK, 2015).
Pathophysiology. ADH is synthesized in the hypothalamus and stored and secreted by the posterior pituitary; it manages the reabsorption of water by the distal tubules and collecting ducts in the kidneys. ADH deficiency leads to excessive diuresis. With all types of DI, patients can urinate from 3-15 L per day. Other signs and symptoms include dry skin, polydipsia (increased thirst), fatigue, dizziness, confusion, and nausea (NIDDK, 2015). This diuresis leads to dehydration, increased serum osmolality (concentrated blood), and hypotension. Increased serum osmolality prompts polydipsia, which typically causes some patients to drink enough fluids to maintain fluid balance, at least initially. If the patient is unconscious or has a defective thirst mechanism, dehydration can occur quickly (Hopper, 2015; Mayo Clinic, 2019a; NIDDK, 2015).
Diagnosis. The diagnosis of DI is initially based on a history of risk factors and symptoms. Infants may present with irritability, crying, weight loss or poor growth, and hyperthermia. Children may report fatigue and exhibit anorexia, enuresis, and a linear growth defect (Khardori, 2020).
A urinalysis should be performed to assess urine specific gravity and glucose to rule out DM (Hopper, 2015). A 24-hour urine is typically ordered to assess urine osmolality and specific gravity, as well as tests for serum electrolytes, glucose, osmolality, and sometimes ADH (Khardori, 2020).
If further confirmation is required, a water deprivation (i.e., Miller-Moses) test may be done. This involves depriving the patient of water for up to 6 hours while assessing body weight and osmolality hourly. In addition to the water deprivation test, an MRI is likely to be ordered to assess for any abnormalities around the hypothalamus or pituitary gland (Khardori, 2020).
Treatment. Treatment will vary according to the type of DI. For severe cases requiring hospitalization, hypotonic intravenous (IV) fluids (i.e., 0.45% normal saline [NS]) should be used to replace intravascular volume without adding sodium. For mild DI, the patient may only need to increase their water intake, especially if the thirst mechanism remains intact. Underlying structural abnormalities, such as tumors in the hypothalamus or pituitary, may be corrected surgically. The primary medication used for the maintenance treatment of central and gestational DI is desmopressin (DDAVP, Stimate), a synthetic analog, to replace the missing ADH and decrease urination. It is available subcutaneous (SQ) or IV, as well as an intranasal spray and an oral tablet. It is dosed BID/TID (NIDDK, 2015). Patients should be educated regarding common adverse effects, including flushing, HA, rhinitis, nausea, abdominal pain, dizziness, and hyponatremia (Khardori, 2020).

Central Hypothyroidism
Thyroid disease (see Figure 2) is generally characterized as overactive (hyperthyroidism) or underactive (hypothyroidism) functioning. Hypothyroidism is classified as primary or secondary; the primary form is much more common. Secondary, or central, hypothyroidism is caused by the failure of the pituitary gland or hypothalamic disease, as the body does not make adequate amounts of TRH/TSH to stimulate the release of T3/4 (see Figure 3). This occurs in approximately 1 in 20,000 to 80,000 people (Ross, 2019a; Shahid et al., 2020). For additional information regarding the pathophysiology, underlying epidemiology, presentation, diagnosis, and management of primary hypothyroidism, explore the NursingCE activity entitled Thyroid Dysfunction, available on NursingCE.com.

Pathophysiology. Secondary hypothyroidism is caused by a failure to stimulate normal thyroid function and not the thyroid itself. Secondary hypothyroidism occurs due to hypothalamus or pituitary disease or trauma, resulting in diminished TRH or TSH secretion (American Thyroid Association [ATA], n.d.a). Central hypothyroidism may also be associated with ADH, oxytocin, PRL, FSH, LH, GH, and ACTH deficiencies (Duke Health, 2018). It may result from a tumor, infection, infarction, or TBI causing damage to the hypothalamus or pituitary gland. Iatrogenic causes involve pituitary/hypothalamus function, such as previous radiation therapy or surgical trauma (Ross, 2019a; Shahid et al., 2020).
Regardless of the cause, if the thyroid gland does not produce enough thyroid hormone, all mental and physical processes slow down. The thyroid manages metabolism, and a slowed metabolic rate is responsible for many of the characteristic symptoms. Furthermore, cellular functions are diminished, reducing oxygen consumption, oxidation of nutrients for energy, and body heat (ATA, n.d.a).
Diagnosis. Symptoms of central hypothyroidism resemble those of primary hypothyroidism—such as fatigue, cold intolerance, muscle cramps, HA, and weight gain—but may be less pronounced (Ross, 2019a). In children, central hypothyroidism may reduce growth velocity (Shahid et al., 2020). Recognizing the signs and symptoms of hypothyroidism can be difficult as they may be vague and nonspecific: for example, extreme fatigue is often the first symptom reported by those suffering from hypothyroidism (ATA, n.d.a). Other symptoms of hypothyroidism in adult patients can include:
- bradycardia
- constipation
- mental dullness
- shortness of breath
- decreased perspiration
- facial edema (especially periocular)
- joint pain
- hair thinning or loss
- dry skin and hair
- sexual dysfunction such as menstrual irregularities/amenorrhea (Hopper, 2015)
In addition to the history and physical examination, a serum TSH level should be performed to establish a diagnosis of central hypothyroidism. According to the US Preventive Services Task Force (2015), TSH is considered the first-line screening test for patients with suspected thyroid dysfunction. The level of circulating TSH in the blood helps determine if the thyroid is functioning normally, overactive, or underactive. T4 can be measured as total T4 or free (FT4). Total T4 measures both the free and the bound hormone available, whereas FT4 assesses the T4 hormone that is freely circulating in the blood and available for use. FT4 is more commonly performed since it provides the best insight into the severity of an abnormal TSH level. FT4 is most accurate when performed in conjunction with a TSH level, so these tests are usually ordered together (ATA, 2019; BCGuidelines, 2018; Snyder, 2020b).
A thyroid panel typically consists of three main tests: TSH, FT4, and free T3 (T3, Free) or total T3 (American Board of Internal Medicine, 2019). Free T3 is less reliable and not clinically indicated in suspected thyroid disease. The total T3 test is reserved for identifying hyperthyroidism or determining its severity, as patients with an overactive thyroid have elevated T3 levels (ATA, 2019).
With secondary hypothyroidism, the TSH level is typically normal or slightly decreased, and the T4 level often decreases (Ross, 2019b). The most common thyroid conditions classified by TSH and FT4 values are demonstrated in Table 2 (ATA, n.d.b).

A complete blood count (CBC) and metabolic profile should also be completed. Patients with hypothyroidism may exhibit:
- anemia;
- hyponatremia;
- hyperlipidemia; and
- elevations of transaminases, creatinine kinase, and alkaline phosphates (Orlander, 2019; Ross, 2019b).
An electrocardiogram (ECG) may be indicated and may demonstrate sinus bradycardia, flat or inverted T-waves, or low-voltage QRS complexes (ATA, n.d.a; Hopper, 2015). In addition, the adrenal function must be fully assessed via a fasting morning serum cortisol level prior to initiating corrective treatment for secondary hypothyroidism (Ross, 2019a). Thyroid scanning using technetium-99m or iodine-123 or an ultrasound can help provide information about the etiology of the disease (Daniel, 2017).
Treatment. Treatment for most cases of hypothyroidism begins with a low dose of levothyroxine (Synthroid), which is a synthetic form of T4. The typical initial dose for an adult with mild to moderate hypothyroidism is 50-75 µg/day (Orlander, 2019). Treatment with glucocorticoids for adrenal insufficiency should be started before or concurrently with thyroid hormone replacement but not after (Snyder, 2019c).
The goal of treatment is to replace the hormone that is no longer being made by the thyroid. Clinical benefits should occur within 5 days of initiating the medication and level off in about 4 weeks (Orlander, 2019). As the patient’s T4 is being replaced by the levothyroxine (Synthroid), their FT4 level should gradually normalize (Ross, 2019a). The primary danger of levothyroxine is taking too much or too little. If too much thyroid hormone is taken, signs and symptoms of hyperthyroidism will result. If too little is taken, the clinical manifestations of hypothyroidism will persist. During annual clinical evaluations, a patient on levothyroxine (Synthroid) therapy should be evaluated for the classic symptoms of hypothyroidism or hyperthyroidism (Orlander, 2019).

Patient education for hypothyroidism should include the following points:
- Thyroid hormone replacement can increase the risk of bleeding with anticoagulation therapy; patients should be taught to monitor for signs and symptoms of bleeding and report any changes immediately.
- Thyroid hormone replacement can interact with digoxin (Lanoxin); patients should monitor their pulse daily.
- Patients should inform all providers of their condition and current medication list to avoid potential interactions with other medications or therapies.
- Patients should avoid the use of herbals or OTC medications without prior approval from their health care team, as they may alter the effects of their thyroid hormone therapy.
- Patients are encouraged to take good care of their skin; hypothyroidism patients typically report dry skin, which may lead to breakdown. They should avoid perfumed soaps or shower gels and moisturize daily with fragrance-free lotion.
- Patients should avoid commercial weight-loss products and maintain a well-balanced diet. Rapid weight loss is a sign of hyperthyroidism and may indicate overtreatment.
- Constipation is common with hypothyroidism, so a high-fiber diet with adequate fluid intake is essential (ATA, n.d.a).
Complications. If not well managed, the potential complications of thyroid dysfunction include:
- bleeding tendencies;
- benign intracranial hypertension;
- atherosclerosis, decreased peripheral circulation, ischemic heart disease, heart failure, cardiomegaly, or other cardiovascular conditions;
- deafness;
- infertility;
- GI disturbances such as pernicious anemia, achlorhydria (lack of hydrochloric acid), megacolon (abnormal dilation of the colon), or GI obstruction;
- iron-deficiency anemia;
- psychiatric symptoms; and
- myxedema coma (ATA, n.d.a; Hopper, 2015).
Myxedema coma usually occurs in patients with long-standing or untreated hypothyroidism. The condition can be triggered by infection, trauma, or exposure to extreme cold. The patient will develop hypothermia (under 95o F), bradypnea (decreased respiratory rate), lethargy, hypoglycemia, decreased cardiac output, and a decreased level of consciousness. Death can ensue as a result of cardiac or respiratory failure if untreated (Hopper, 2015).
By rapidly diagnosing myxedema coma, treatment can be expedited. Myxedema coma requires aggressive treatment due to its life-threatening nature. The ATA makes a strong recommendation based on low-quality evidence that parenteral glucocorticoid therapy should be given at stress doses for the initial treatment of myxedema coma, prior to levothyroxine (Synthroid) administration. Parenteral levothyroxine (Synthroid) is typically administered. Once the patient has improved clinically, they should receive oral doses of levothyroxine (Synthroid) according to their TSH/FT4 level. Additionally, liothyronine (Cytomel) may be given in addition to levothyroxine (Synthroid; Jonklaas et al., 2014).
Gonadotropic Dysfunction
Amenorrhea, or the lack of menstruation in females, can either be defined as primary or secondary. Primary amenorrhea is defined as no menses by age 15, although it may also be diagnosed at age 13 in the context of no secondary sexual characteristics (Welt & Barbieri, 2020a). Secondary amenorrhea is clinically defined as a lack of menses for 3 or more months in a woman whose menstrual cycles were previously regular, or 6 or more months in a woman whose cycles were irregular previously (Shahid et al., 2020; Welt & Barbieri, 2020b).
Primary Hypogonadism
Primary hypogonadism refers to a pathology affecting the ovaries or testes themselves, not the entire hypothalamic-pituitary-gonadal (HPG) axis. In women, this may present as POI; in men, it may present as infertility. In either sex, primary hypogonadism may also involve delayed puberty. Delayed puberty is defined in the US as a lack of secondary sexual characteristics by the age of 12 in females and the age of 14 in males (Crowley & Pitteloud, 2018). The symptoms of hypogonadism are relatively consistent, whether the pathology is primary or secondary. Hypogonadism in females—primarily due to reduced estrogen and progesterone levels—typically presents with menstrual irregularities, including oligomenorrhea, amenorrhea, and anovulation. Reduced estrogen levels can also cause hot flashes, night sweats, insomnia, vaginal tissue atrophy, decreased libido, mood swings, dry skin, decreased BMD, and dyspareunia in women. Elevated levels of estrogen in females may lead to weight gain, dysmenorrhea (more painful menstruation), menorrhagia (heavier) or lighter menstrual periods, worsened mood swings, bloating, and breast tenderness immediately prior to menstruation (premenstrual syndrome, or PMS), fibrocystic breasts, uterine fibroid development, fatigue, decreased libido, and symptoms of depression or anxiety (El Sayed et al., 2020; Hormone Health Network [HHN], 2018, 2019; Shahid et al., 2020). Hypogonadism in men—typically related to testosterone deficiency—can lead to decreased libido, erectile dysfunction, decreased sperm count, and gynecomastia. It can cause reduced nocturnal penile tumescence (spontaneous nocturnal or morning erections), loss of body hair, decreased BMD, smaller testes, fatigue, depression, anemia, increased body fat, and reduced muscle mass. Chronically, low testosterone can lead to decreased bone density, decreased energy, and infertility. Elevated testosterone levels may lead to premature puberty in adolescents. Estrogen deficiency in males may prompt an increase in abdominal adiposity and decreased libido, while estrogen levels that are higher than expected in men may lead to gynecomastia, erectile dysfunction, and infertility (El Sayed et al., 2020; HHN, 2018, 2019, 2020; Shahid et al., 2020; Snyder, 2020d).
Pathophysiology. POI is characterized by ovarian failure with hypogonadism despite elevated levels of FSH. About 75-90% of cases are categorized as idiopathic with no known cause, while autoimmune disease accounts for roughly 4% of cases. Other less common causes of POI include iatrogenic injury (e.g., chemotherapy and radiation therapy), Turner syndrome, fragile X, and other genetic defects (Welt, 2018).
Primary hypogonadism in males damages the seminiferous tubules, making sperm production and fertility not possible (Snyder, 2019b). Potential causes of primary hypogonadism include genetic defects (Klinefelter syndrome), accidental testicular injury, or iatrogenic injury due to chemotherapy, radiation therapy, surgery, and infection (Surampudi et al., 2014).
Delayed puberty may be categorized as either primary or secondary. Primary (hypergonadotropic) hypogonadism accounts for 13% of delayed puberty cases. It may be due to Turner syndrome, Klinefelter syndrome, an autoimmune disorder, infection, cryptorchidism, deficiency of testosterone biosynthesis, or iatrogenic causes, such as chemotherapy or radiation treatment (Crowley & Pitteloud, 2018). Gonadal dysgenesis—a lack of gonadal development—is responsible for as much as 43% of primary amenorrhea cases; another 15% may be related to Mullerian agenesis, or a lack of vaginal development (Welt & Barbieri, 2020a).
Diagnosis. The signs and symptoms of primary hypogonadism, including POI, mirror those of other forms of hypogonadism due to estrogen, progesterone, and testosterone deficiency (Snyder, 2020a). A thorough history and physical exam should provide helpful information. The physical examination should include an evaluation of the anatomy of the uterus, cervix, and vagina. Breast development, height, weight, and pubic hair presence or distribution should be noted to establish an accurate Tanner stage for both male and female patients. Acne or hirsutism should be noted (Welt & Barbieri, 2020a). POI typically manifests before the age of 40, differentiating itself from the natural course of menopause. In 50-75% of cases, intermittent ovarian function occurs, potentially leading to irregular menses (Welt, 2019).
No additional laboratory testing is required for women presenting with regular menses. In cases of amenorrhea, a pregnancy test should be performed first to rule out the possibility of pregnancy. An estradiol level that is below the expected reference range, in combination with LH/FSH levels that are above expected, indicates primary hypogonadism. (Corenblum, 2020; Fleseriu et al., 2016; Snyder, 2020a, 2020b). Laboratory studies in patients with POI typically demonstrate an elevated FSH level. An FSH level in the postmenopausal range per the laboratory reference is diagnostic of overt POI, while an FSH level above 10-15 IU/L and serum estradiol of 80 pg/mL or greater indicate occult POI or a diminishing ovarian reserve (Welt, 2019).
Physical examination in males should note the penile and testicular anatomy, size, and any abnormal findings, especially signs of hypogonadism. Testing in males should be performed in the absence of acute/subacute illness and before 10 a.m. following an overnight fast. A reduced serum testosterone level confirms hypogonadism in males, while an elevated LH indicates primary hypogonadism (Corenblum, 2020; Fleseriu et al., 2016; Snyder, 2020a, 2020b, 2020d).
The primary presenting signs of delayed puberty among both males and females involve a lack of secondary sexual characteristics. Recommended laboratory tests include testosterone (males), estradiol (females), LH, FSH, PRL, TSH, and FT4. Tests should be performed before 10 a.m. after an overnight fast (Crowley & Pitteloud, 2018). As previously stated, an abnormally low estradiol level in a female patient in combination with an elevated LH/FSH level indicates primary hypogonadism. A reduced serum testosterone level confirms hypogonadism in men; an elevated LH indicates primary hypogonadism (Snyder, 2020b).
Treatment. Estrogen replacement in POI patients has been shown to reduce the risk of osteopenia and cardiovascular disease and relieve the symptoms of urogenital atrophy. As with other forms of hypogonadism, treatment varies by age and fertility concerns. Younger girls (ages 11-12) should be started on a low-dose estrogen 17-beta transdermal patch (Vivelle). After 24 months, 200 mg of cyclic micronized progesterone (Prometrium) can be added on menstrual cycle days 1-12. The treatment of secondary amenorrhea due to POI differs slightly. If seeking fertility, these patients should be counseled that only 5-10% of patients with POI achieve pregnancy, and this is not the primary goal of treatment. They can be started on 100 mcg/day of estradiol, given either transdermal via a patch (Vivelle) or the vaginal ring (NuvaRing). If the uterus remains intact, progesterone should be administered to avoid endometrial hyperplasia. In patients with secondary amenorrhea who are not seeking fertility, a combined oral contraceptive (ethinyl estradiol and progestin [Loestrin]) of choice can be utilized (Welt, 2020).
The goal of treatment for men with primary infertility does not involve reestablishing spermatogenesis, as this is often not possible. However, treatment may provide the benefits of virilization, improved sexual function, increased muscle mass, decreased body fat, improved BMD, and potentially improved mood, although this final benefit remains uncertain. Testosterone replacement may increase the patient's cardiovascular risk slightly, but this is unclear currently. Testosterone is the primary treatment option for primary hypogonadism in men. Current topical options in the US include three gel preparations of testosterone (Androgel 1% or 1.6%, Testim 1%, and Fortesta 2%) and one solution (Axiron 2%). All must be applied daily to varying locations. Patients using Androgel should be cautioned regarding skin irritation, although this has been reported with all topical formulas. Patients who are prescribed Testim may report an unpleasant odor. All topical applications carry a risk of transferring the medication to family members and other close contacts. This may pose a significant risk for pregnant partners or infants and small children. Patients should be instructed to practice diligent hand hygiene after applying the medication, allow the medication to dry fully after application, and avoid getting the medicated body part wet for at least 5 hours after application (Snyder, 2020d; Surampudi et al., 2014).
A testosterone patch (Androderm) is also available. Unfortunately, up to 30% of users report a severe skin rash with this option. Injectable options in the US include testosterone enanthate (Delatestryl), which is administered intramuscularly (IM) every 1-2 weeks. Testosterone undecanoate (Aveed) is injected via deep IM (gluteus) less frequently. This version is difficult to access and is currently being monitored by the federal Risk Evaluation and Mitigation Strategy (REMS) program due to the risk of pulmonary oil microembolism and anaphylaxis (Snyder, 2020d; Surampudi et al., 2014).
A weekly SQ injectable testosterone enanthate (Xyosted) was approved for use in the US in recent years, but clinically it is not used frequently due to insufficient evidence regarding its safety and efficacy at this time. Some oral preparations are 17-alpha alkylated to slow metabolism by the liver. A buccal testosterone tablet (Striant SR) is dosed BID and meant to be dissolved along the upper anterior gum line. Implantable SQ pellets of testosterone (Testopel) are implanted in the subcutaneous tissue of the buttocks, abdomen, or thigh every 3-6 months. Patients should be warned of the risks of extrusion, infection, or fibrosis when using implantable pellets. A nasal spray preparation of testosterone (Natesto) is also available in the US. It should be dosed TID, which may decrease patient compliance but carries less risk of treatment transfer to close contacts. Most users (73%) report rhinorrhea, epistaxis, sinusitis, or nasal scabbing. Any formulation of testosterone can cause acne, gynecomastia, weight gain, fluid retention, increased HDL, benign prostatic hyperplasia, increased hemoglobin/hematocrit/red blood cells, and sleep apnea. Testosterone may also increase the risk of prostate cancer, and treatment is contraindicated in those with a history of prostate or breast cancer (Snyder, 2020d; Surampudi et al., 2014).
Testosterone treatment is contraindicated for patients with lower urinary tract symptoms, erythrocytosis (hematocrit above 50%), severe untreated sleep apnea, or uncontrolled congestive heart failure. Testosterone and LH levels should be monitored. Injectable hCG is not used for the treatment of primary hypogonadism but is discussed in greater detail in the forthcoming section on the treatment of secondary hypogonadism in men seeking fertility (Snyder, 2020d; Surampudi et al., 2014).
The treatment of females with delayed puberty should follow the recommendations for POI in young girls with primary amenorrhea; if required, their treatment should gradually transition to adult dosages as above. Young males should be treated with testosterone, which is typically dosed as 50 mg of testosterone enanthate (Delatestryl) or cypionate (Depo-Testosterone) monthly. In the US, testosterone gels are not approved for use in patients under the age of 18, and there are no oral preparations available for this age group. If long-term treatment is required for male patients, treatment with testosterone is most common, as fertility is not a treatment goal among these patients (Crowley & Pitteloud, 2018).
Secondary Hypogonadism
Secondary hypogonadism—also referred to as central or hypogonadotropic hypogonadism—is a lack of LH and FSH production by the anterior pituitary, which leads to deficiencies in estrogen, progesterone, and/or testosterone (Snyder, 2020a).
Pathophysiology. The underlying causes of secondary hypogonadism mirror those previously listed for other forms of hypopituitarism (Snyder, 2019a). These can include infection, infiltrative disease, tumor, radiation therapy, traumatic injury, empty sella, or infarct/apoplexy. Hyperprolactinemia may also cause amenorrhea that is either primary or secondary (Welt & Barbieri, 2020a).
Functional hypothalamic amenorrhea (FHA) is a form of secondary hypogonadism caused by hypothalamic dysfunction; it is believed to be responsible for approximately 35% of all cases of secondary amenorrhea and 2% of cases of primary amenorrhea. FHA is characterized by a reduction in the secretion of gonadotropin-releasing hormone (GnRH), which thereby diminishes the secretion of LH and FSH by the anterior pituitary and the secretion of estrogen and progesterone by the ovaries. Additionally, it may cause a reduction in IGF-1 and an increase in cortisol levels; this is potentially due to a diversion of limited energy resources to systems that are prioritized over the reproductive system (Shahid et al., 2020).
Secondary, or hypogonadotropic hypogonadism, accounts for the vast majority of delayed puberty cases. CDGP is the underlying cause in 53% of cases of secondary hypogonadism in children and adolescents, while 19% of cases are due to FHA. The remaining 12% of cases are due to other less-common etiologies, including isolated GnRH deficiency. CDGP is a familial disorder defined as a transient functional defect in GnRH secretion by the hypothalamus, and most cases are temporary and resolve spontaneously with time and treatment. In contrast, only 10% of isolated cases of GnRH deficiency resolve spontaneously (Crowley & Pitteloud, 2018).
Diagnosis. The signs and symptoms of secondary hypogonadism mirror those of other forms of hypogonadism due to estrogen, progesterone, and testosterone deficiency related to any cause (Snyder, 2020a). Other indications of hypothalamus or pituitary dysfunction include HA, visual changes, fatigue, polyuria, and polydipsia. A thorough history may indicate the underlying pathology responsible for hypogonadism. For example, a history of neonatal crisis may indicate congenital adrenal hyperplasia; poor childhood health may implicate hypothalamic/pituitary disease; acne and hirsutism may indicate polycystic ovarian syndrome (PCOS); recent stress, a change in diet, or a change in exercise routine may indicate FHA; and galactorrhea may indicate hyperprolactinemia. Finally, illicit drug use—especially heroin and methadone—may lead to amenorrhea in chronic users (Welt & Barbieri, 2020a). Risk factors for FHA include decreased body weight (BMI below 18), anorexia nervosa, excessive exercise, and emotional distress, all of which typically lead to an imbalance in energy intake and output, creating a prolonged energy deficit. In addition to amenorrhea, patients with FHA will typically exhibit decreased BMD, mood disorders, and other signs of estrogen deficiency like breast tissue and vaginal atrophy, and dyspareunia or other indications of sexual dysfunction (Shahid et al., 2020).
A pregnancy test should be done to rule out the possibility of pregnancy first. Estradiol, LH, and FSH levels that are all below the expected reference ranges indicate secondary hypogonadism in a female patient. Testing for central hypogonadism should occur in the absence of acute/subacute illness and before 10 a.m. following an overnight fast. In males, a reduced serum testosterone level confirms hypogonadism, and a reduced or normal LH indicates secondary hypogonadism (Corenblum, 2020; Fleseriu et al., 2016; Snyder, 2020a, 2020b).
Bone age should be assessed in those diagnosed with delayed puberty, as patients with CDGP typically present with a bone age that is 20% younger than their chronological age. MRI may help assess for any space-occupying lesions that could be disrupting the patient's normal endocrine function (Crowley & Pitteloud, 2018).
Treatment. FHA should be treated using a multidisciplinary approach, including behavioral therapy with psychological and/or psychiatric support if appropriate. Treatment goals should include weight gain and stress reduction. Patients with congenital GnRH deficiency should be referred to an endocrine practice with experience managing this condition but are typically treated using exogenous gonadotropins or pulsatile GnRH (not currently available in the US commercially, only as a research-based therapy). These patients should also be referred to a reproductive endocrinologist if fertility is desired (Welt & Barbieri, 2020a, 2020b).
Treatment for secondary hypogonadism varies based on sex and the fertility concerns of the patient. Premenopausal females who are not seeking fertility should be treated with a combination of transdermal estradiol (Vivelle) daily with progesterone (Prometrium, Provera). Premenopausal females who are seeking fertility should be treated with gonadotropins, LH, and FSH (Novarel, Menopur) via injection, the details of which are outside the scope of this activity. Postmenopausal females should be treated symptomatically only with estradiol and progestin (Premarin; Snyder, 2019c).
Males who are not seeking fertility can be treated with exogenous testosterone as described above for primary hypogonadism. Males seeking fertility should be treated with gonadotropins for pituitary disease or GnRH or gonadotropins for hypothalamus disease (Mayo Clinic, 2020; Snyder, 2019c). Fertility restoration is more likely to be effective in males if their hypogonadism is mild to moderate, the condition began after puberty was completed, the patient has bilateral descended testicles, and the patient has a prior history of treatment with testosterone or gonadotropins. First-line treatment should include hCG (Novarel) to replace the deficient LH, as it is less expensive and more likely to be effective as a monotherapy compared to FSH. The sperm count should be rechecked every 1-3 months. If the sperm count remains below 5 million/mL after 6 months of combined hCG/hMG treatment, the dose of hMG can be increased. While the sperm count may rise as quickly as 6-10 months after starting treatment, pregnancy typically takes 21-38 months. Alternative treatments that are appropriate for patients with hypothalamic deficiency only include pulsatile GnRH and clomiphene citrate (Clomid; Snyder, 2019b).
Treatment for patients with delayed puberty due to secondary hypogonadism mirrors the previously described treatments above for primary delayed puberty (Crowley & Pitteloud, 2018).
For learners who are eager to access additional content related to the endocrine system and endocrine disorders, please see the following NursingCE courses:
- Endocrine and Hormonal Disorders Part 1: Anatomy and Physiology
- Endocrine and Hormonal Disorders Part 3: Syndromes of Excessive Hormone Secretion
- Endocrine and Hormonal Disorders Part 4: Adrenal Gland Disorders
- Diabetes
- Osteoporosis
- Thyroid Dysfunction
- Sexual Dysfunction
References
American Board of Internal Medicine. (2019). ABIM laboratory test reference ranges - January2019. https://www.abim.org/~/media/ABIM%20Public/Files/pdf/exam/laboratory-reference-ranges.pdf
American Thyroid Association. (n.d.a). Hypothyroidism (underactive). Retrieved July 16, 2020, from https://www.thyroid.org/hypothyroidism/
American Thyroid Association. (n.d.b). Thyroid function tests. Retrieved May 29, 2020, from https://www.thyroid.org/thyroid-function-tests/
American Thyroid Association. (2019). Thyroid function tests FAQ. https://www.thyroid.org/wp-content/uploads/patients/brochures/thyroid_function_tests_faq.pdf
BCGuidelines. (2018). Thyroid function testing in the diagnosis and monitoring of thyroid function disorder. https://www2.gov.bc.ca/assets/gov/health/practitioner-pro/bc-guidelines/thyroid-function-testing.pdf
CFCF. (2014). Thyroid and parathyroid. [Image]. https://commons.wikimedia.org/wiki/File:Illu_thyroid_parathyroid.jpg
Chapman, I. M. (2019). Gigantism and acromegaly. In Merck Manual of diagnosis and therapy. Merck Sharp & Dohme. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/gigantism-and-acromegaly.
Cleveland Clinic. (2020). Somatropin, rh-GH injection. https://my.clevelandclinic.org/health/drugs/19180-somatropin-rh-gh-injection
Corenblum, B. (2020). Hypopituitarism (panhypopituitarism). https://emedicine.medscape.com/article/122287-overview
Crowley, W. F., & Pitteloud, N. (2018). Approach to the patient with delayed puberty. UpToDate. https://www.uptodate.com/contents/approach-to-the-patient-with-delayed-puberty
Daniel, M. S. (2017). Congenital hypothyroidism. https://emedicine.medscape.com/article/919758-overview#a1
Duke Health. (2018). Thyroid disorders in children. https://www.dukehealth.org/pediatric-treatments/pediatric-endocrinology/thyroid-disorders-children
El Sayed, S. A., Fahmy, M. W., & Schwartz, J. (2020). Physiology, pituitary gland. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK459247/
Fleseriu, M., Hashim, I. A., Karavitaki, N., Melmed, S., Murad, M. H., Salvatori, R., & Samuels, M. H. (2016). Hormonal replacement in hypopituitarism in adults: An endocrine society clinical practice guideline, The Journal of Clinical Endocrinology & Metabolism, 101(11), 3888–3921. https://doi.org/10.1210/jc.2016-2118
Gounden, V., & Jialal, I. (2020). Hypopituitarism (panhypopituitarism). StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK470414/
Häggström, M. (2009). Thyroid hormones [image]. https://commons.wikimedia.org/wiki/File:Thyroid_system.svg
Hopper, P. D. (2015). Understanding medical-surgical nursing (5th ed.). FA Davis.
Hormone Health Network. (2018). Estrogen. https://www.hormone.org/your-health-and-hormones/glands-and-hormones-a-to-z/hormones/estrogen
Hormone Health Network. (2019). Pituitary gland. https://www.hormone.org/your-health-and-hormones/glands-and-hormones-a-to-z/glands/pituitary-gland
Hormone Health Network. (2020). Testosterone and androgens. https://www.hormone.org/your-health-and-hormones/glands-and-hormones-a-to-z/hormones/testosterone
Ignatavicius, D. D., Workman, M. L., Rebar, C. R., & Heimgartner, N. M. (2018). Medical-surgical nursing concepts for interprofessional collaborative care (9th ed.). Elsevier.
Jonklaas, J., Bianco, A. C., Bauer, A. J., Burman, K. D., Cappola, A. R., Francesco, S. C., Cooper, D. S., Kim, B. W., Peeters, R. P., Rosenthal, M. S., & Sawka, A. M. (2014). Guidelines for the treatment of hypothyroidism. Thyroid, 24(12). https://doi.org/10.1089/thy.2014.0028
Khardori, R. (2020). Diabetes insipidus. https://emedicine.medscape.com/article/117648-overview
Mayo Clinic. (2018). Dwarfism. https://www.mayoclinic.org/diseases-conditions/dwarfism/symptoms-causes/syc-20371969
Mayo Clinic. (2019a). Diabetes insipidus. https://www.mayoclinic.org/diseases-conditions/diabetes-insipidus/symptoms-causes/syc-20351269
Mayo Clinic. (2019b). Hypopituitarism. https://www.mayoclinic.org/diseases-conditions/hypopituitarism/symptoms-causes/syc-20351645
Mayo Clinic. (2020). Follicle-stimulating hormone and luteinizing hormone (intramuscular route, subcutaneous route). https://www.mayoclinic.org/drugs-supplements/follicle-stimulating-hormone-and-luteinizing-hormone-intramuscular-route-subcutaneous-route/description/drg-20062932
National Institute of Diabetes and Digestive and Kidney Diseases. (2015). Diabetes insipidus. https://www.niddk.nih.gov/health-information/kidney-disease/diabetes-insipidus
OpenStax College. (2013). The endocrine system. [Image]. https://commons.wikimedia.org/wiki/File:1801_The_Endocrine_System.jpg
Orlander, P. R. (2019). Hypothyroidism. https://emedicine.medscape.com/article/122393-overview
Richmond, E. J., & Rogol, A. D. (2020a). Diagnosis of growth hormone deficiency in children. UpToDate. https://www.uptodate.com/contents/diagnosis-of-growth-hormone-deficiency-in-children
Richmond, E. J., & Rogol, A. D. (2020b). Diagnostic approach to children and adolescents with short stature. UpToDate. https://www.uptodate.com/contents/diagnostic-approach-to-children-and-adolescents-with-short-stature
Rogol, A. D., & Richmond, E. J. (2020). Treatment of growth hormone deficiency in children. UpToDate. https://www.uptodate.com/contents/treatment-of-growth-hormone-deficiency-in-children
Ross, D. S. (2019a). Central hypothyroidism. UpToDate. https://www.uptodate.com/contents/central-hypothyroidism
Ross, D. S. (2019b). Diagnosis of and screening for hypothyroidism in nonpregnant adults. UpToDate. https://www.uptodate.com/contents/diagnosis-of-and-screening-for-hypothyroidism-in-nonpregnant-adults
Shahid, Z., Asuka, E., & Singh, G. (2020). Physiology, hypothalamus. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK535380/
Snyder, P. J. (2019a). Causes of hypopituitarism. UpToDate. https://www.uptodate.com/contents/causes-of-hypopituitarism
Snyder, P. J. (2019b). Induction of fertility in men with secondary hypogonadism. UpToDate. https://www.uptodate.com/contents/induction-of-fertility-in-men-with-secondary-hypogonadism
Snyder, P. J. (2019c). Treatment of hypopituitarism. UpToDate. https://www.uptodate.com/contents/treatment-of-hypopituitarism
Snyder, P. J. (2020a). Clinical manifestations of hypopituitarism. UpToDate. https://www.uptodate.com/contents/clinical-manifestations-of-hypopituitarism
Snyder, P. J. (2020b). Diagnostic testing for hypopituitarism. UpToDate. https://www.uptodate.com/contents/diagnostic-testing-for-hypopituitarism
Snyder, P. J. (2020c). Growth hormone deficiency in adults. UpToDate. https://www.uptodate.com/contents/growth-hormone-deficiency-in-adults
Snyder, P. J. (2020d). Testosterone treatment of male hypogonadism. UpToDate. https://www.uptodate.com/contents/testosterone-treatment-of-male-hypogonadism
Society for Endocrinology. (2018). Pituitary apoplexy. https://www.yourhormones.info/endocrine-conditions/pituitary-apoplexy/
Surampudi, P., Swerdloff, R. S., & Wang, C. (2014). An update on male hypogonadism therapy. Expert Opinion on Pharmacotherapy, 15(9), 1247–1264. https://doi.org/10.1517/14656566.2014.913022
US Preventive Services Task Force. (2015). Screening for thyroid dysfunction: Recommendation statement. American Family Physician, 91(11), 790A-790F. https://www.aafp.org/afp/2015/0601/od1.pdf
Welt, C. K. (2018). Pathogenesis and causes of spontaneous primary ovarian insufficiency (premature ovarian failure). UpToDate. https://www.uptodate.com/contents/pathogenesis-and-causes-of-spontaneous-primary-ovarian-insufficiency-premature-ovarian-failure
Welt, C. K. (2019). Clinical manifestations and diagnosis of spontaneous primary ovarian insufficiency (premature ovarian failure). UpToDate. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-spontaneous-primary-ovarian-insufficiency-premature-ovarian-failure
Welt, C. K. (2020). Management of spontaneous primary ovarian insufficiency (premature ovarian failure). UpToDate. https://www.uptodate.com/contents/management-of-spontaneous-primary-ovarian-insufficiency-premature-ovarian-failure
Welt, C. K., & Barbieri, R. L (2020a). Evaluation and management of primary amenorrhea. UpToDate. https://www.uptodate.com/contents/evaluation-and-management-of-primary-amenorrhea
Welt, C. K., & Barbieri, R. L (2020b). Evaluation and management of secondary amenorrhea. UpToDate. https://www.uptodate.com/contents/evaluation-and-management-of-secondary-amenorrhea
Willis, L. M. (2019). Professional guide to diseases (11th ed.). Wolters Kluwer.