The purpose of this module is to provide the APRN with an overview of the pathophysiology, risk factors, clinical presentation, diagnostic workup, evidence-based management guidelines, and potential complications of primary endocrine disorders leading to hormonal excess, including acromegaly, gigantism, hyperprolactinemia, syndrome of inappropriate antidiuretic hormone secretion, polycystic ovarian syndrome, hyperparathyroidism, and multiple endocrine neoplasia type 1.
...purchase below to continue the course
location that precludes surgical removal, then radiation or other treatments may be indicated (Chapman, 2019).
Radiation may be recommended if part of the tumor remains after surgery or in cases of inoperable tumors. Radiation will destroy lingering tumor cells and help reduce GH levels. The following types of radiation may be used as therapy for acromegaly:
- Conventional radiation therapy is given every weekday over 4-6 weeks but could take up to 10 years to realize the full effect.
- Proton beam therapy delivers a targeted, high dose of radiation to the tumor and spares normal surrounding tissue.
- Stereotactic radiosurgery delivers a high dose of radiation to tumor cells in a single dose. It limits the amount of radiation exposure to normal surrounding tissue and may normalize GH levels within 5 years (Mayo Clinic, 2019).
Unfortunately, iatrogenic hypopituitarism may develop several years after radiation therapy (Chapman, 2019). Medications that lower or block GH production should be considered for patients who are not surgical candidates, cannot obtain radiation treatment, have completed surgery or radiation therapy without success, or are awaiting surgery or radiation therapy to start or take full effect. These medications are as follows:
- Octreotide (Sandostatin), octreotide acetate (Sandostatin LAR), and lanreotide (Somatuline Depot) are somatostatin analogs, prompting a decrease in the secretion of GH by the pituitary. Octreotide (Sandostatin) is dosed at 50-150 µg SQ every 8-12 hours, while octreotide acetate (Sandostatin LAR) is dosed at 10-40 mg IM every 4-6 weeks. Sandostatin LAR is only recommended for those who were previously stable/tolerating the SQ formula of Sandostatin. Lanreotide (Somatuline Depot) should be dosed at 60-120 mg SQ every 4 weeks.
- The dopamine agonists cabergoline (Dostinex) and bromocriptine (Parlodel) decrease levels of GH and IGF-1 less effectively than the somatostatin analogs. Nausea, postural hypotension, and mental fogginess are common side effects of both of these medications. Less commonly, both agents can cause an adverse effect of compulsive behaviors such as gambling during administration and should be monitored closely. Cabergoline (Dostinex) has been associated with valvular heart disease at higher doses.
- Pegvisomant (Somavert) is a new GH receptor antagonist that can be administered as a daily injection if other treatments are not effective. This drug can normalize IGF-1 levels and relieve symptoms, but it does not lower GH levels or reduce tumor size (Chapman, 2019; Mayo Clinic, 2019; Snyder, 2020b).
The APRN should educate patients in the following areas:
- instruction on reducing stress as much as possible prior to the GH suppression test, as stress can artificially elevate GH levels;
- instruction on the treatment regime, ensuring the patient and their family understand the importance of compliance;
- alerting the patient and their family regarding the dangers of OTC drugs, herbals, and other medications that may interact with prescribed treatment;
- instruction to wear a medical alert bracelet to alert caregivers of diagnosis; and
- annual follow-up with labs and imaging studies, if appropriate, is crucial for managing acromegaly and avoiding long-term and potentially life-threatening complications (Mayo Clinic, 2019).
Hyperprolactinemia
Prolactin (PRL) is a hormone produced by lactotrophs in the anterior pituitary. It targets the gonads and mammary glands by binding to peptide hormone receptors. It manages the growth and development of mammary glands in pubescent girls, as well as milk production in postpartum women. Its secretion is regulated by the hypothalamus, predominantly through inhibition (via the secretion of PIH, or dopamine), as well as PRH and TRH, which increase secretion to a lesser degree (El Sayed et al., 2020).
Pathophysiology
Hyperprolactinemia, or an excess of circulating PRL, is often caused by a PRL-secreting adenoma (termed a prolactinoma), medications, pregnancy, lactation, increased stress, or cranial radiation therapy (El Sayed et al., 2020). It may also be caused by hypothalamic dysfunction, specifically a reduction in the secretion of dopamine (Shahid et al., 2020). Providers should review the patient’s current list of medications to ensure they are not taking any agents that may increase serum PRL concentrations, such as neuroleptic drugs (e.g., fluphenazine [Prolixin], haloperidol [Haldol], paliperidone [Invega], risperidone [Risperdal], clomipramine [Anafranil], as well as other first-generation antipsychotic medications), metoclopramide (Reglan), methyldopa (Aldomet), and cimetidine (Tagamet; Snyder, 2020b). An elevation in the PRL level inhibits the secretion of GnRH by the hypothalamus, luteinizing hormone/follicle-stimulating hormone (LH/FSH) by the anterior pituitary, and eventually estrogen/progesterone by the ovaries, leading to anovulation, amenorrhea, and other menstrual irregularities (El Sayed et al., 2020).
Diagnosis
Due to the secondary decrease in estrogen and progesterone, the most common signs and symptoms of hyperprolactinemia in premenopausal women include hypogonadism, as well as occasional galactorrhea and HA. In men, hyperprolactinemia may cause decreased libido, erectile dysfunction, infertility, gynecomastia/galactorrhea, and other signs of hypogonadism (Snyder, 2020a). Postmenopausal women are typically asymptomatic, as they are hypogonadotropic at baseline (Shahid et al., 2020). Since secretion is episodic, more than one abnormal PRL level is typically required for diagnosis. An elevated fasting morning serum PRL level above 20 ng/mL (20 µg/L) in men and postmenopausal women or above 30 ng/mL (20 µg/L) in premenopausal women is typically considered diagnostic. As isolated hyperprolactinemia is less common, providers should consider the concurrent evaluation of LH, FSH, thyroid-stimulating hormone (TSH), and free T4 (FT4) levels to assess for other indications of endocrine dysfunction (Snyder, 2020a).
Treatment
If medications are believed to be the underlying cause, they should be adjusted when possible. In the event of a lactotroph adenoma, a surgical consult is recommended to determine whether the lesion can be removed surgically, particularly for patients who are not responding to or cannot tolerate pharmacological treatment. As previously mentioned, surgery is typically performed via transsphenoidal resection and is the preferred treatment option for adenomas measuring greater than 3 cm at diagnosis, especially in women seeking fertility. Lactotroph microadenomas (under 1 cm) and macroadenomas under 3 cm in size should be treated pharmacologically with a dopamine agonist such as cabergoline (Dostinex) or bromocriptine (Parlodel). Cabergoline has been more effective in inducing menstruation and facilitating pregnancy with fewer side effects, although it does carry an increased risk of valvular heart disease at higher doses. An echocardiogram is recommended every 2 years for patients taking more than 2 mg weekly. Dosing for cabergoline (Dostinex) should start at 0.25 mg twice weekly and be increased to the lowest effective dose in order to limit this risk. The maximum dosage recommended is 1.5 mg two to three times weekly. As mentioned above, both medications involve side effects of nausea, postural hypotension, and mental fogginess. Less frequently, these agents can cause an adverse effect of compulsive behaviors. Nausea is significantly more common with bromocriptine (Parlodel), but QHS dosing may limit this effect with both medications. Bromocriptine (Parlodel) should be prescribed at a starting dose of 1.25 mg QHS for the first week; if tolerated, it can be increased to BID dosing during the second week. The maximum dosage recommended is 5 mg BID. Alternatives for women who do not respond to or cannot tolerate treatment with dopamine agonist therapy and are not seeking fertility include exogenous estradiol and progesterone therapy. These patients can be offered treatment with combined oral contraceptives (COCs) or separate low-dose estradiol and progesterone. Similarly, men with hyperprolactinemia who do not respond to or cannot tolerate treatment with dopamine agonist therapy can receive exogenous testosterone if they are not seeking fertility; human chorionic gonadotropin (hCG [Novaerl]) may be considered if the patient seeks fertility. For additional information on the treatment of hypogonadism, refer to the next section (Snyder, 2020b).
For patients with microadenomas, the serum PRL level should be rechecked monthly while on pharmacologic therapy until they are stable and then at least yearly. After a year of stable dosing and serum PRL levels, the provider is typically encouraged to decrease the dose. If the patient remains stable on the medication for at least 2 years, an MRI should be repeated to ensure the resolution of the adenoma. If the MRI is negative (i.e., no evidence of adenoma on imaging) and the patient has been stable for at least 2 years, the gradual discontinuation of medication can be considered. Medication can also be discontinued after menopause in most women (Snyder, 2020b).
For patients with a macroadenoma (greater than 1 cm in size) who are on dopamine agonist therapy, imaging should be repeated to assess for a decrease in the size of the lesion at 6-12 months. The serum PRL level should be rechecked monthly initially until this stabilizes, at 6 months, and then at least yearly. After a year of stable dosing/serum PRL levels, the provider is typically encouraged to decrease the dose. If the patient remains stable on the medication for at least 2 years and the MRI results are negative, the gradual discontinuation of the medication can be considered. However, the provider should consider continuing therapy (if well tolerated) for patients with macroadenomas that are greater than 2 cm in size at the time of diagnosis. As previously mentioned, a surgical consult is recommended for any patient who is not responding to pharmacological treatment. Postoperative radiation therapy may be indicated to prevent regrowth after the surgical debulking of larger macroadenomas that are resistant to dopamine agonist therapy (Snyder, 2020b).
Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH)
The hypersecretion of antidiuretic hormone (ADH) leads to a reduction in water secretion by the kidneys, inducing hyponatremia, a decrease in serum osmolality, and an increase in urine osmolality (Thomas, 2019).
Pathophysiology
Increased secretion of ADH is typically related to a central nervous system disorder, such as a stroke, hemorrhage, infection, traumatic brain injury, malignancy, or medications. The medications most often associated with increased ADH secretion include chlorpropamide (Diabinese), carbamazepine (Tegretol), cyclophosphamide (Cytoxan), and selective serotonin reuptake inhibitors (SSRIs; Shahid et al., 2020).
Diagnosis
SIADH typically presents with signs of hyponatremia, such as nausea or vomiting, HAs, difficulty thinking, weakness, and restlessness (Shahid et al., 2020). Patients may also present with ataxia or falls. Patients with chronic disease may present asymptomatically or appear minimally symptomatic. The classic diagnostic criteria by Bartter and Schwartz that were developed in 1967 are still applicable. They include the following:
- serum hyponatremia combined with hypoosmolality
- continued renal excretion of sodium (urine sodium above 40 mEq/L)
- urine less than maximally dilute
- lack of clinical evidence of volume depletion
- no other obvious causes of hyponatremia
- a correction/improvement in hyponatremia with fluid restriction (Thomas, 2019)
Other causes of hyponatremia consist of adrenal insufficiency (mineralocorticoid deficiency, glucocorticoid deficiency), hypothyroidism, congestive heart failure (CHF), hypopituitarism, renal disease with salt wastage, hepatic disease, and drugs that impair renal water excretion. The differential diagnoses for SIADH include volume depletion or dehydration, CHF, and liver cirrhosis. Physical examination findings that indicate hypovolemia should decrease the level of suspicion for SIADH (e.g., hypotension, orthostasis, dry mucous membranes, cold peripheries, decreased skin turgor). Liver cirrhosis and CHF typically cause hypervolemia, as evidenced by peripheral edema, increased jugular venous pressure, pulmonary rales, or ascites. Laboratory evaluation of SIADH patients typically indicates serum sodium below 135 mmol/L, serum osmolality below 280 mOsm/kg, and urine osmolality above 100 mOsm/kg. Serum uric acid should be assessed—it is typically reduced to below 4 mg/dL—and most experts recommend assessing the patient’s cortisol, TSH, and plasma arginine vasopressin (AVP). Providers should expect a reduction in the anion gap, a reduced BUN, and an increased glomerular filtration rate (GFR) due to volume expansion. Advanced imaging studies are not always indicated for SIADH patients, but they may show a space-occupying lesion or cerebral edema, as evidenced by the narrowing of the ventricles (Thomas, 2019).
Treatment
Hyponatremia related to SIADH should be corrected slowly (max 8-12 mEq/L in the first 24 hours) to avoid osmotic demyelination syndrome, which is also known as central pontine myelinolysis (CPM; Shahid et al., 2020; Thomas, 2019). The primary goal should be to correct the patient’s sodium by 0.5-1 mEq/L per hour, with a maximum of 125-130 mEq/L. The rapidity of treatment should be adjusted based on the severity and chronicity of the condition. One benchmark termed the “Rule of Sixes” suggests that the patient’s sodium level should be increased by 6 mEq/L over the first 24 hours or by 6 mEq/L over the first 6 hours and then stopped. The recommendations according to the American Expert Panel include:
- limiting the rate of correction if the condition is chronic;
- initiating fluid restriction as the first line of treatment, limiting intake to 500-1500 mL/day;
- considering pharmacological treatment only if serum sodium levels remain abnormally low after 24-48 hours of fluid restriction;
- avoiding fluid restrictions for patients on vasopressin-2 receptor antagonists (aquaretics or vaptans); and
- utilizing water, 5% dextrose, or desmopressin to slow the rate of correction if diuresis is profound (Thomas, 2019).
More aggressive management should occur for patients presenting with more severe symptoms of hyponatremia (e.g., seizures, a decreased level of consciousness, and respiratory arrest) or in cases involving acute moderate to severe hyponatremia (less than 48 hours in duration). This may include the infusion of 3% hypertonic saline solution. The treatment team should monitor the patient closely during this time for any changes in neurological status, in addition to regular monitoring of the serum sodium level. Sodium deficiency should be calculated using the patient’s serum sodium level as well as weight and then used to determine the volume of solution required based on a 0.5-mEq/L rate of correction, which is the appropriate infusion rate. Alternative treatment options for acute sodium correction include the use of loop diuretics (e.g., furosemide [Lasix]) in combination with normal saline or 3% and V2 receptor antagonists. The same close monitoring techniques noted earlier are recommended for these patients. When treating chronic cases of SIADH, fluid restriction is the preferred first-line treatment. Medication, typically a V2 receptor antagonist (aquaretic or vaptan), should be considered if the patient’s sodium level does not increase by at least 2 mEq/L after 24 hours of a 1,000-mL fluid restriction. Conivaptan (Vaprisol) is only approved for use in hospitalized patients in an IV formula. Tolvaptan (Samsca) is available as an oral formula for the treatment of hypervolemic or euvolemic hyponatremia. Initial dosing should start at 15 mg daily, titrating up carefully to a max of 60 mg daily with no fluid restriction in place while titrating the dose. If the patient is asymptomatic and stable, they can be discharged after the first 24-48 hours. Patients can remain on tolvaptan (Samsca) long term if needed, or the medication can be tapered carefully a few weeks after the resolution of an underlying cause for SIADH, such as tumor removal, resolution of infection, or discontinuation of the implicated medication. The patient’s sodium level should be monitored carefully during the taper and for at least 5 days following discontinuation. Alternative medications for the treatment of chronic SIADH include loop diuretics or urea. Loop diuretics help increase the excretion of free water by inhibiting sodium and chloride resorption in the kidneys, while urea helps increase urine volume by increasing the urinary osmotic or solute load. It should be dosed based on weight (0.5 g/kg) and comes packaged as a powder that should be dissolved in water for convenient administration. Urea should be avoided in patients with creatinine over 2 mg/dL or a BUN of 80 mg/DL or higher (Thomas, 2019).
Hyperparathyroidism
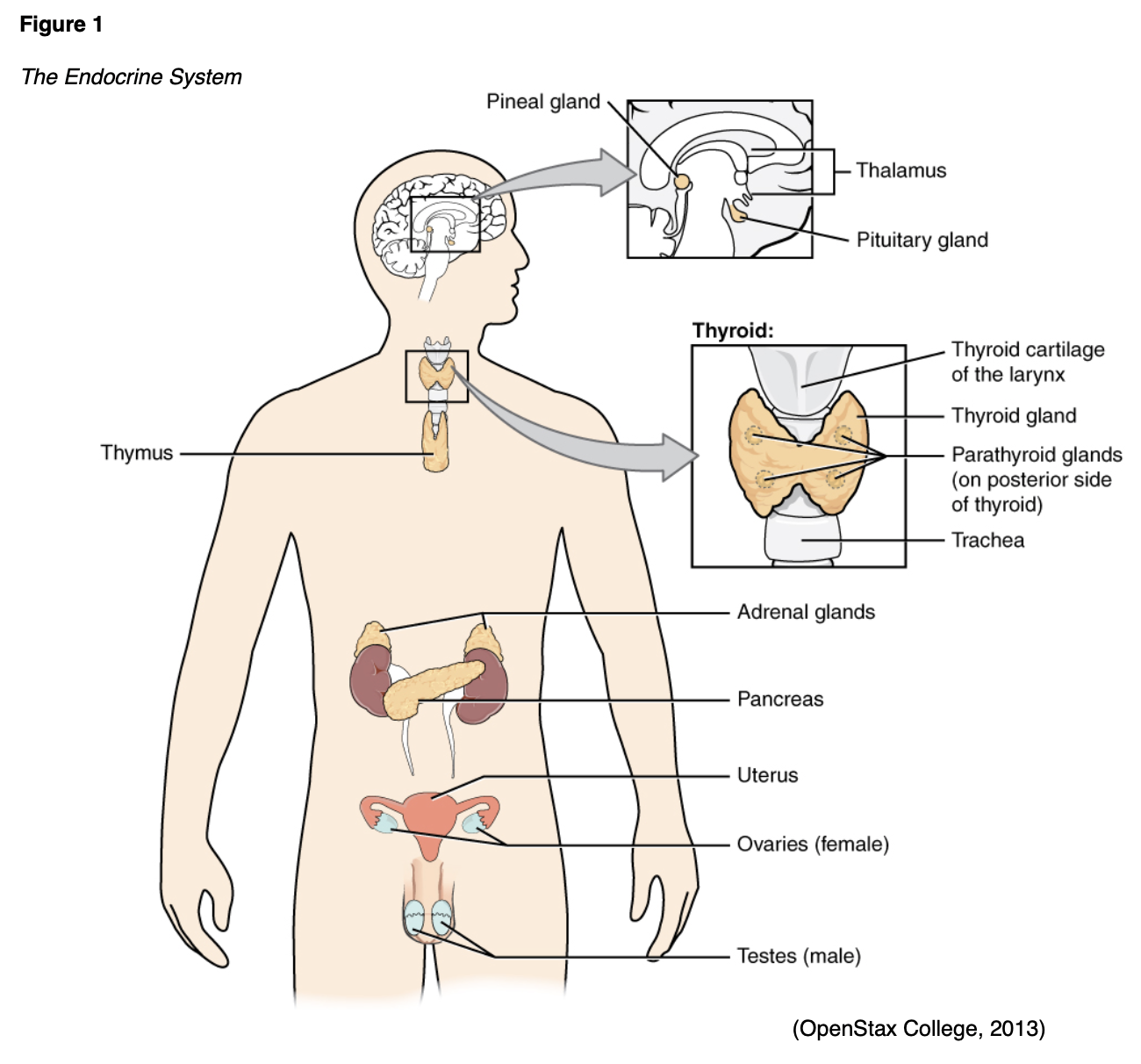
The parathyroid glands are four small glands located just on the posterior surface of the thyroid gland (see Figures 1 and 4). As previously mentioned, these structures regulate circulating serum calcium and phosphorus via the secretion of PTH. While hypocalcemia related to a deficiency in PTH is possible, it is exceedingly rare. More commonly, an excess of PTH leads to hypercalcemia, which increases the risk of nephrolithiasis (kidney stones), osteopenia, and eventually fractures (National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK], 2019).
Pathophysiology
Hyperparathyroidism may be primary (related to parathyroid gland pathology) or secondary to chronic kidney disease (CKD), chronic vitamin D deficiency, or altered absorption (e.g., inflammatory bowel disease). In the US, the incidence of primary hyperparathyroidism is approximately 100,000 new cases annually, with a prevalence of 1 in 600 to 1,000 people. Approximately 80% of these cases are caused by a benign adenoma in one of the parathyroid glands. A smaller portion (10-15%) of primary hyperparathyroidism patients are diagnosed with hyperplasia, in which all four glands are enlarged. Rarely, primary hyperparathyroidism may be caused by MEN1, familial hypocalciuric hypercalcemia, or malignancy of the parathyroid gland. Risk factors include age between 50 and 60, and the condition affects roughly three to four times more women than men. It is most common in patients with African American ethnicity, followed by Caucasian patients (NIDDK, 2019).
Diagnosis
Most patients with hyperparathyroidism are asymptomatic. Symptoms may be mild and include muscle weakness, fatigue, depression, and painful joints/bones. Patients with more severe hyperparathyroidism may present with confusion, decreased appetite, nausea/vomiting, constipation, polyuria, and polydipsia. The diagnosis of hyperparathyroidism is typically made incidentally on routine laboratory testing. Patients with increased serum calcium should be further evaluated, and the diagnosis is typically confirmed by an elevated serum PTH level, elevated calcium (greater than 400 mg/d) on a 24-hour urine collection, and creatinine clearance below 60 mL/min. These patients should also undergo a DEXA scan to assess their BMD for possible osteopenia and a serum 25 hydroxyvitamin D test to assess for deficiency. Imaging studies—a CT scan, an ultrasound, or a sestamibi scan (using a parenteral radioactive tracer)—may be warranted to rule out nephrolithiasis and gather additional information for surgical planning (NIDDK, 2019).
Treatment
The first-line treatment for primary hyperparathyroidism is the surgical excision of the affected gland (typically one of four). Surgical indications include:
- serum calcium level greater than 1 mg/dL above the expected normal range,
- BMD per DEXA scan below -2.5 at any site,
- history or evidence of nephrolithiasis,
- fragility fracture (e.g., related to a ground-level fall), or
- age over 50 years (NIDDK, 2019).
A focused or minimally invasive parathyroidectomy is usually performed as an outpatient or same-day procedure. Research indicates that surgical excision leads to both improved BMD and a reduced risk of nephrolithiasis. For parathyroid hyperplasia, surgery typically involves the excision of three and a half of the patient's four parathyroid glands via bilateral neck exploration. Patients who are not surgical candidates or opt to treat their condition more conservatively may be treated with cinacalcet (Sensipar) to reduce serum calcium levels. Cinacalcet (Sensipar) works by increasing the sensitivity of the parathyroid gland’s calcium-sensing receptors (CaSRs) and therefore reducing the secretion of PTH. Cinacalcet has not been shown to improve BMD in patients with primary hyperparathyroidism, and patients being treated with cinacalcet should also receive counseling on the potential benefits of a bisphosphonate to improve BMD if fracture risk is a concern. Vitamin D should also be supplemented daily if the patient’s serum 25 hydroxyvitamin D is below the normal range (NIDDK, 2019).
Polycystic Ovary Syndrome
PCOS is the most common endocrine disorder affecting females during their reproductive years (Teede et al., 2018). PCOS affects 8-20% of females, many of whom are undiagnosed. About one in 10 females with PCOS will develop type 2 DM (T2DM), and 30-40% will develop impaired glucose tolerance by age 40 years; approximately 70% of ovulatory and fertility issues among premenopausal women is due to PCOS. There are no known causes of PCOS, but risk factors include genetics, insulin resistance, and low-grade inflammation that may lead to the disorder. Females who have a first-degree relative with PCOS (i.e., mother or sister) are more likely to develop the disorder than those without this family history. Several health risks are associated with PCOS. These include:
- insulin resistance and eventually T2DM;
- cardiac disease;
- sleep apnea;
- abnormal uterine bleeding;
- obesity that can lead to low self-esteem;
- metabolic syndrome, including HTN, hyperlipidemia, elevated BG, and increased waist circumference;
- steatohepatitis (non-alcoholic fatty liver);
- depression and anxiety; and
- endometrial cancer (Lucidi, 2019; Smith, 2018; Teede et al., 2018).
Patients with PCOS report a decreased quality of life, and the condition is associated with an increased risk of gestational DM, pregnancy-induced HTN (PIH), miscarriage, myocardial infarction (MI), and endometrial cancer (Smith, 2018; Teede et al., 2018).
Pathophysiology
An imbalance of reproductive hormones characterizes PCOS (see Figure 10). This hormonal imbalance leads to ovarian dysfunction. The ovaries normally release one egg each month during ovulation, as part of the menstrual cycle. In patients with PCOS, the egg may not develop completely or may not be released during ovulation. PCOS can prompt metrorrhagia (irregular menses) or amenorrhea, which can lead to infertility (Office on Women’s Health [OWH], 2019). While the pathophysiology of PCOS is not completely understood, it is characterized by relatively high levels of androgens (i.e., testosterone, androstenedione, and DHEA), which is likely due to an overproduction of LH by the anterior pituitary. An increase in LH leads to relatively low FSH and decreased production of estrogen by the ovaries. This imbalance may be related to an abnormality in cytochrome P450c17, the rate-limiting enzyme in androgen biosynthesis, or the regulation of CYP17 (Lucidi, 2019).

Anovulation in patients with PCOS is likely due to the high levels of androgens, which can also cause excess hair growth and acne—both signs of PCOS. As patients with PCOS age, their menstrual cycles typically become increasingly regular as they near menopause; however, the hormonal imbalance does not improve with age and the other symptoms of PCOS persist. While the risk of type 2 DM, MI, and stroke increases with age among all women, the risk is even higher in women with PCOS (OWH, 2019).
Diagnosis
PCOS should be suspected if symptoms of hyperandrogenism appear, including acne, alopecia (hair loss on the scalp), hirsutism (extra hair growth on the face, chin, or body), and metrorrhagia. Metrorrhagia is an expected finding within the first year following menarche, after which it is defined as cycles under 21 days or over 45 days for women within 3 years of menarche, and cycles under 21 days or over 35 days for women who have been menstruating for more than 3 years (Teede et al., 2018). The physical exam should also include:
- BP;
- height, weight, and body mass index (BMI);
- waist circumference;
- assessment for other symptoms of endocrine disorders; and
- a pelvic exam to assess for signs of high androgen levels such as an enlarged clitoris and enlarged ovaries (OWH, 2019; Teede et al., 2018).
An hCG level should be drawn to rule out pregnancy for women with amenorrhea. Regular menses may occur despite anovulation, and this rare combination should be confirmed by performing a serum progesterone level. Blood tests for androgen levels can include free androgen index, calculated free testosterone, or calculated bioavailable testosterone using a high-quality assay, such as a liquid chromatography-mass spectrometry (LCMS). Free testosterone testing is more sensitive but may be less reliable than total testosterone (Teede et al., 2018).
The Rotterdam criteria are still the most widely utilized clinical diagnostic criteria for the diagnosis of PCOS in adults (Teede et al., 2018). The Rotterdam criteria include three qualifiers, two of which must be present for PCOS diagnosis: androgen excess, ovulatory dysfunction, and polycystic ovaries (Lucidi, 2019). Clinical guidelines recommend the use of transvaginal ultrasound to diagnose polycystic ovaries in sexually active women who consent but do not recommend this method for patients who are still within 8 years of menarche due to the high rate of multifollicular ovaries in this age group. However, a diagnosis of PCOS can be made based on the presence of convincing signs and symptoms of hyperandrogenism in combination with irregular menses (Teede et al., 2018). While the diagnostic criteria for adolescents are not as clear, hyperandrogenemia (elevated systemic levels of androgens) is essential for the diagnosis in this age group (Lucidi, 2019).
LH and FSH levels should be assessed to rule out ovarian failure. For women with regular menstrual cycles, the labs should be drawn between days 5 and 9 of their cycle. (Lucidi, 2019; OWH, 2019: Teede et al., 2018). Typically, a patient with PCOS will have a relatively elevated LH level for their Tanner stage, sex, or age; an FSH level that is normal or slightly below normal; and an LH:FSH ratio that is above 3. Differential diagnoses for patients with suspected PCOS include:
- hyperprolactinemia,
- ovarian/stromal hyperthecosis (hyperplasia of the theca interna of the ovary),
- late-onset congenital adrenal hyperplasia,
- medication-induced hyperandrogenism (due to anabolic corticosteroids, danazol [Danocrine], or androgenic progestins),
- hypothyroidism,
- idiopathic or familial hirsutism,
- CS due to adrenal carcinoma (presenting with elevated androgens, hypokalemia, central obesity, elevated cortisol, and striae), and
- masculinizing tumors of the adrenal gland or ovary (Lucidi, 2019).
Patients diagnosed with PCOS should undergo cardiovascular disease risk evaluation, including a lipid profile for patients who are overweight, as well as glucose-tolerance testing using a reliable assessment method such as a 75-g oral glucose tolerance test (OGTT), fasting plasma glucose, or hemoglobin A1c (HbA1c; Teede et al., 2018). Thyroid function should be assessed using TSH and potentially FT4, and a fasting serum PRL is typically recommended. If there are any concerns regarding adrenal function, cortisol testing should be completed; the American Association of Family Physicians recommends cortisol screening for PCOS patients. If there are any concerns regarding GH dysfunction, an IGF-1 level should also be considered (Lucidi, 2019).
Treatment
Treatment for PCOS is multifactorial. It is important to discuss options for symptomatic control, the patient’s plans for having children, and the risks of long-term problems such as T2DM or heart disease. Goals should be set for symptom control, and the plan of care should be developed around those goals. Furthermore, referrals to a dietician for weight management, an endocrinologist, and reproductive specialists may be indicated depending on the severity of the patient’s PCOS. First-line treatment should consist of lifestyle changes such as exercise and diet (Lucidi, 2019). At least 150 minutes of moderate-intensity exercise per week is recommended, and behavioral therapy/programs may help achieve a 5-10% weight reduction in patients who are overweight or those with central obesity or insulin resistance (Teede et al., 2018).
Pharmacological treatment options for those with hyperandrogenism and menstrual irregularities include combined oral contraceptives (COCs) with 20 mcg ethinyl estradiol and progestin (norethindrone [Loestrin, Junel, Microgestin]). A formulation with ferrous fumarate (Lo Loestrin Fe) may be used for patients with mild anemia. If lifestyle modifications and COCs are not effective, 1500 mg daily of metformin (Glucophage) may be tried, especially for those patients with impaired glucose tolerance. An antiandrogen (aromatase inhibitor) treatment such as letrozole (Femara) may be considered for the management of hirsutism that persists despite at least 6 months of COC and cosmetic treatments (Teede et al., 2018). The Endocrine Society's 2013 practice guidelines concur that adolescents with PCOS and hyperandrogenism should be managed with hormonal contraceptives and metformin (Glucophage). The guidelines further consider oral contraceptives as the first-line treatment for menstrual abnormalities and hirsutism or acne. For females who have a significantly elevated mean platelet volume (MPV), which is associated with adverse cardiovascular events, the use of metformin (Glucophage) is indicated to reduce the MPV and decrease risk (Lucidi, 2019).
For patients with PCOS who are seeking fertility, ovulation-induction agents may be trialed, but the patient should first be informed that these medications are currently considered off-label. This includes the aromatase inhibitor letrozole (Femara), metformin (Glucophage), and clomiphene citrate (Clomid; Teede et al., 2018). The Endocrine Society's 2013 practice guidelines list the first-line treatment for infertility as clomiphene (Clomid; Lucidi, 2019). Pregnancy should be ruled out prior to starting any of these medications to improve fertility, and metformin (Glucophage) and clomiphene citrate (Clomid) can be combined for greater efficacy if needed. Letrozole (Femara) should be dosed at 2.5-5 mg PO and given daily on cycle days 8 through 12. If these medications are ineffective, exogenous gonadotropins could be considered to induce fertility, although ultrasound monitoring is recommended, and the patient should be counseled extensively regarding the cost of the medications and the increased risk for multiple embryos or ovarian hyperstimulation syndrome (OHSS). These medications may be given with or without metformin (Glucophage). Laparoscopic ovarian surgery could also be considered a second-line treatment for clomiphene citrate (Clomid)-resistant patients with only PCOS and anovulatory infertility. More invasive reproductive procedures, such as in-vitro fertilization, may be considered if these options have been unsuccessful (Teede et al., 2018). Statins may be indicated for patients with dyslipidemia (Lucidi, 2019).
Patient education should include the importance of consistent follow-ups, as PCOS is a lifelong disease with many complications. Early intervention for untoward sequelae should be the goal. The patient should receive education regarding the risks of pregnancy for those who conceive, including gestational DM, PIH, C-section delivery, preterm and post-term delivery, newborns who are large for gestational age, and stillbirth (Lucidi, 2019).
Multiple Endocrine Neoplasia Type 1 (MEN1)
This rare (prevalence 1 in 30,000) endocrine disorder most often affects the parathyroid gland, the pancreas, and/or the pituitary gland. It is typically familial and based on a genetic defect that is inherited in an autosomal dominant pattern, although as many as 10% of cases may be due to a random mutation (NIDDK, 2020).
Pathophysiology
MEN1 is characterized by multiple benign tumors located throughout the endocrine and gastrointestinal systems. Most patients with MEN1 initially present with signs and symptoms of hyperparathyroidism in their early 20s, but the average age of diagnosis is not until the fifth decade of life. The vast majority (95%) of MEN1 patients develop parathyroid tumor(s) by age 50, while 40% develop tumors in the pancreas, duodenum, or another gastrointestinal organ (gastrinoma or insulinoma), and 33% develop an anterior pituitary tumor (prolactinoma). Initially, these tumors cause hypersecretion of the gland's hormone(s); if the tumor(s) grow significantly, it/they may eventually cause hyposecretion (NIDDK, 2020).
Diagnosis
The presenting signs and symptoms of MEN1 mimic those previously discussed throughout this module and vary depending on the organs affected (parathyroid gland versus anterior pituitary) and the severity/stage of the disease (hypersecretion initially, followed by hyposecretion during later stages). In 2012, the diagnostic criteria for MEN1 were last updated to include positive findings of at least one of the following three criteria:
- the presence of two or more MEN1-related tumors,
- the presence of at least one MEN1-related tumor and a first-degree relative with MEN1, or
- the presence of the MEN1 mutation, as determined by genetic testing (NIDDK, 2020).
Genetic testing should be offered and is recommended for any patients presenting with a first-degree relative who has been diagnosed with MEN1, or any patient with a history of two or more benign endocrine or gastrointestinal tumors (NIDDK, 2020).
Treatment
Any patients with suspected MEN1 should be referred to an endocrinologist with experience managing MEN1 cases. Treatment plans for patients with MEN1 typically include regular laboratory monitoring of serum calcium, PTH, PRL, and gastrin. Advanced imaging studies, including MRIs or CT scans, are typically performed to assess for tumors throughout the endocrine and gastrointestinal organs. If possible, surgical procedures to excise or debulk tumors will be performed, similar to treatment options listed above for other benign endocrine tumors. The medications listed above may be used for patients who are not surgical candidates, such as cinacalcet (Sensipar, for parathyroid tumors), cabergoline (Dostinex, a dopamine agonist that can treat prolactinomas), or proton pump inhibitors (PPIs, which can treat a gastrinoma; NIDDK, 2020).
For learners who are eager to access additional content related to the endocrine system and endocrine disorders, please see the following NursingCE courses:
- Endocrine and Hormonal Disorders Part 1: Anatomy and Physiology
- Endocrine and Hormonal Disorders Part 2: Hypopituitarism and Other Hormone Deficiencies
- Endocrine and Hormonal Disorders Part 4: Adrenal Gland Disorders
- Diabetes
- Osteoporosis
- Thyroid Dysfunction
- Sexual Dysfunction
References
BruceBlaus. (2017). Polycystic ovary syndrome. [Image]. https://commons.wikimedia.org/wiki/File:PCOS_(Part_2).png
Chapman, I. M. (2019). Gigantism and acromegaly. In Merck Manual of diagnosis and therapy. Merck Sharp & Dohme. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/gigantism-and-acromegaly.
El Sayed, S. A., Fahmy, M. W., & Schwartz, J. (2020). Physiology, pituitary gland. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK459247/
Hopper, P. D. (2015). Understanding medical-surgical nursing (5th ed.). FA Davis.
Ignatavicius, D. D., Workman, M. L., Rebar, C. R., & Heimgartner, N. M. (2018). Medical-surgical nursing concepts for interprofessional collaborative care (9th ed.). Elsevier.
Lucidi, R. S. (2019). Polycystic ovarian syndrome. https://emedicine.medscape.com/article/256806-overview
Mayo Clinic. (2019). Acromegaly. https://www.mayoclinic.org/diseases-conditions/acromegaly/symptoms-causes/syc-20351222
National Institute of Diabetes and Digestive and Kidney Diseases. (2019). Primary hyperparathyroidism. https://www.niddk.nih.gov/health-information/endocrine-diseases/primary-hyperparathyroidism
National Institute of Diabetes and Digestive and Kidney Diseases. (2020). Multiple endocrine neoplasia type 1. https://www.niddk.nih.gov/health-information/endocrine-diseases/multiple-endocrine-neoplasia-type-1
Office on Women's Health. (2019). Polycystic ovary syndrome. https://www.womenshealth.gov/a-z-topics/polycystic-ovary-syndrome
OpenStax College. (2013). The endocrine system. [Image]. https://commons.wikimedia.org/wiki/File:1801_The_Endocrine_System.jpg
Schwartz, R. A. (2020). Gigantism and acromegaly. https://emedicine.medscape.com/article/925446-overview
Shahid, Z., Asuka, E., & Singh, G. (2020). Physiology, hypothalamus. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK535380/
Smith, L. (2018). What is polycystic ovary syndrome? https://www.medicalnewstoday.com/articles/265309
Snyder, P. J. (2020a). Clinical manifestations and evaluation of hyperprolactinemia. UpToDate. https://www.uptodate.com/contents/clinical-manifestations-and-evaluation-of-hyperprolactinemia
Snyder, P. J. (2020b). Management of hyperprolactinemia. UpToDate. https://www.uptodate.com/contents/management-of-hyperprolactinemia
Teede, H. J., Misso, M. L., Costello, M. F., Dokras, A., Laven, J., Moran, L., Piltonen, T., Norman, R. J., & International PCOS Network. (2018). Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Human reproduction (Oxford, England), 33(9), 1602–1618. https://doi.org/10.1093/humrep/dey256
Thomas, C. P. (2019). Syndrome of inappropriate antidiuretic hormone secretion (SIADH). https://emedicine.medscape.com/article/246650-overview